



Neutrophilic dermatoses (ND) encompass a heterogeneous set of skin diseases, some of which have a chronic and recurrent course. These diseases pose a therapeutic challenge; many different drugs have been used to treat ND, with variable results.1
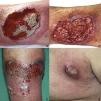
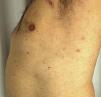
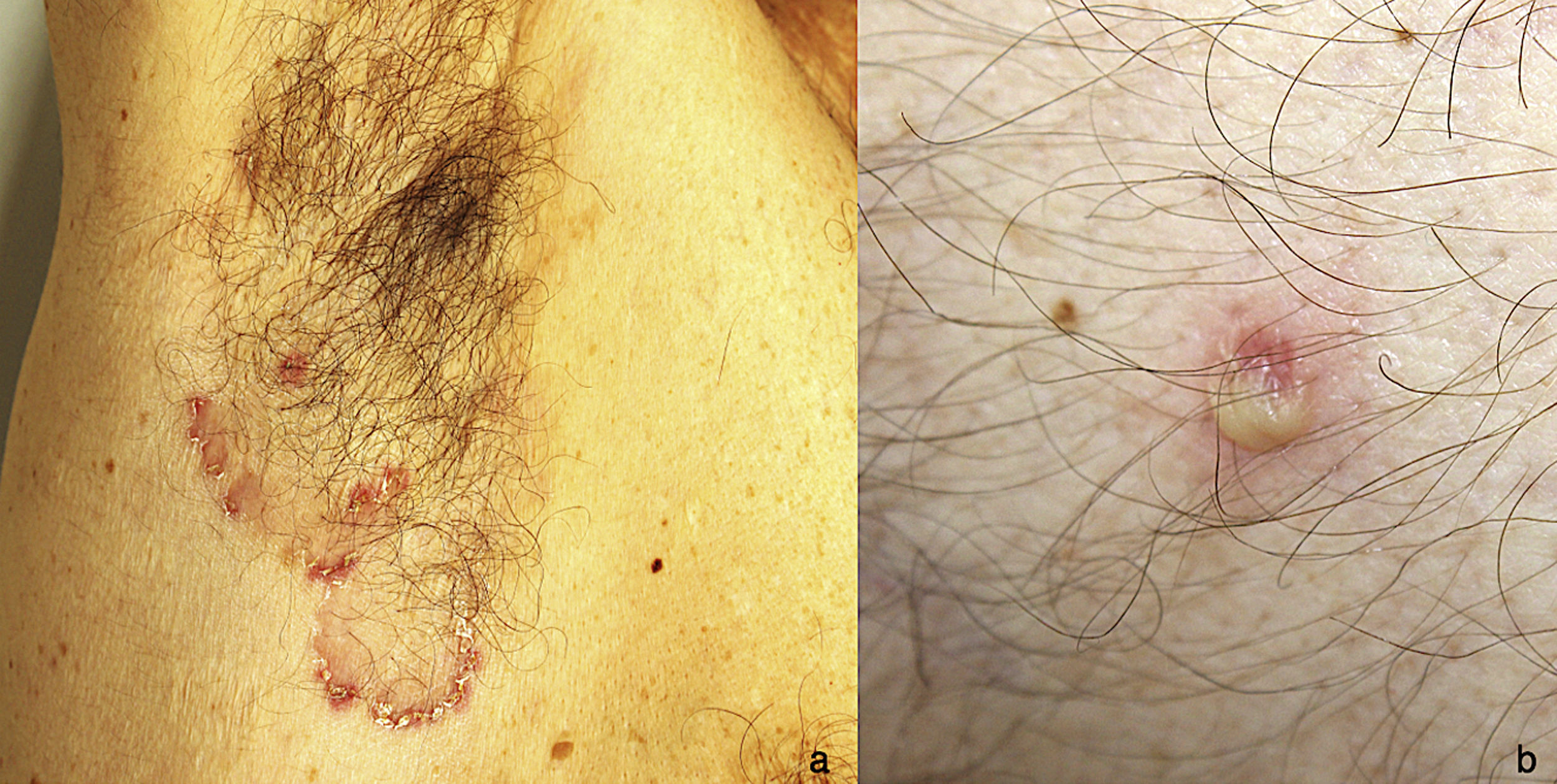
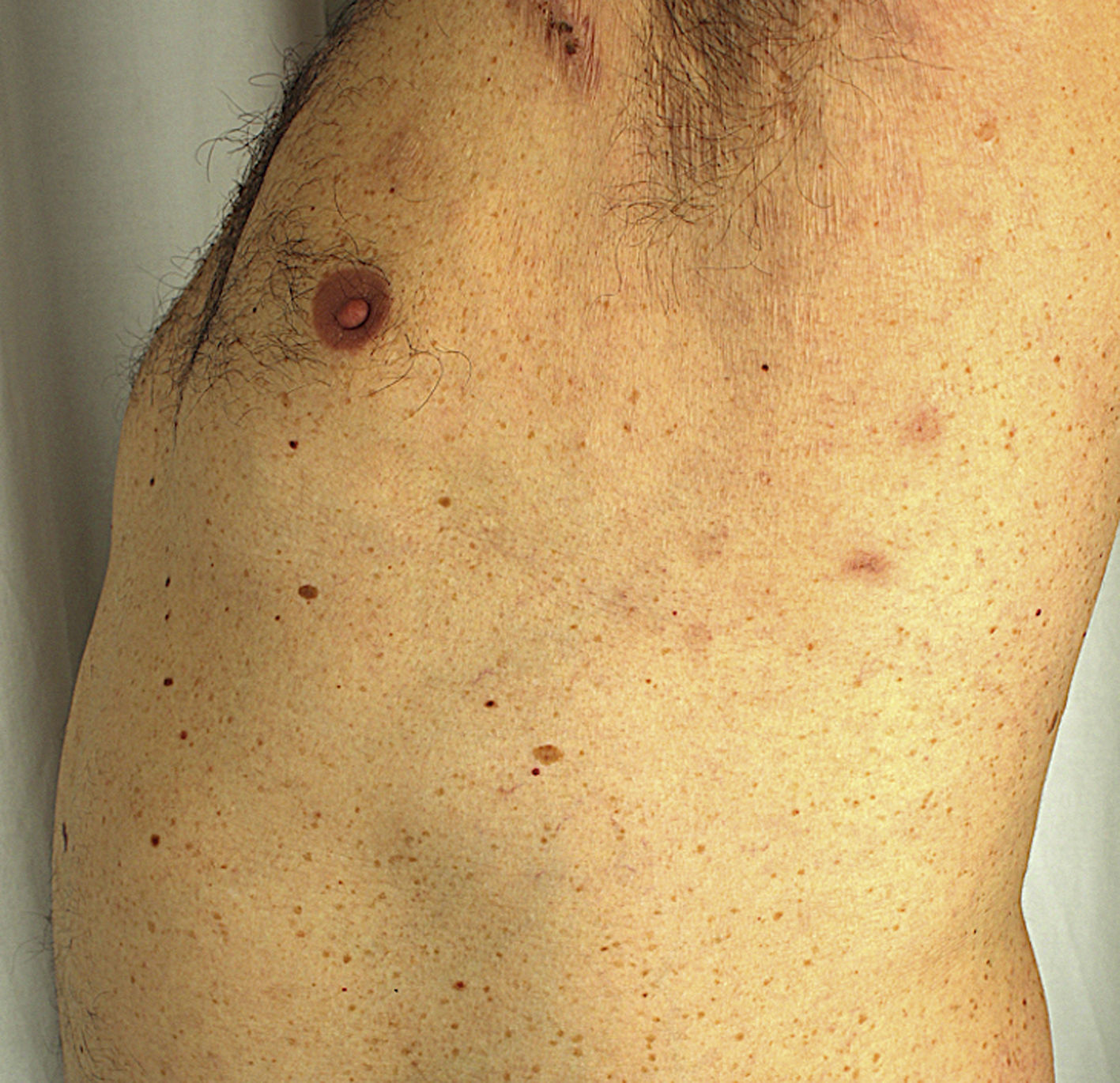
A 51-year-old man with no past medical history of interest was in follow up for recurrent episodes of pyoderma gangrenosum (PG) triggered by trauma, insect bites, or infection (Fig. 1). He reported no systemic symptoms nor any family history of skin disease. Monoclonal immunoglobulin (Ig) A gammopathy of undetermined significance (MGUS) had been detected at the moment of diagnosis, but had not required treatment. PG flare-ups were treated with pulses of oral corticosteroids, as well as topical and intralesional corticosteroids, cyclosporine (up to a maximum dose of 5 mg/kg/d), and topical tacrolimus. While the patient showed a good initial response to these treatments, he experienced rapid recurrences affecting varying locations. Later, the patient developed new lesions in the form of flaccid pustules grouped on the edges of polycyclic erythematous plaques, located on the trunk and armpits (Fig. 2). Culture did not reveal the presence of microorganisms. Histology was compatible with a diagnosis of subcorneal pustular dermatosis (SPD). The patient began treatment with sulfone (100–200 mg/d) and, later, with colchicine (0.5–1 mg/d), with little improvement. He experienced 7 episodes of PG in 1 year, despite various therapeutic attempts and avoidance of triggers. Finally, the patient began treatment with subcutaneous adalimumab (induction dose, 80 mg in wk 0 and 40 mg in wk 1; maintenance dose, 40 mg every 2 wk). The SPD lesions disappeared after administration of the second dose of adalimumab, and no recurrences were observed after 21 months of follow-up (Fig. 3). Episodes of PG were limited to a single new lesion that appeared 6 months after starting treatment and was easily controlled with topical corticosteroid therapy. No further progression of the MGUS has been detected to date.
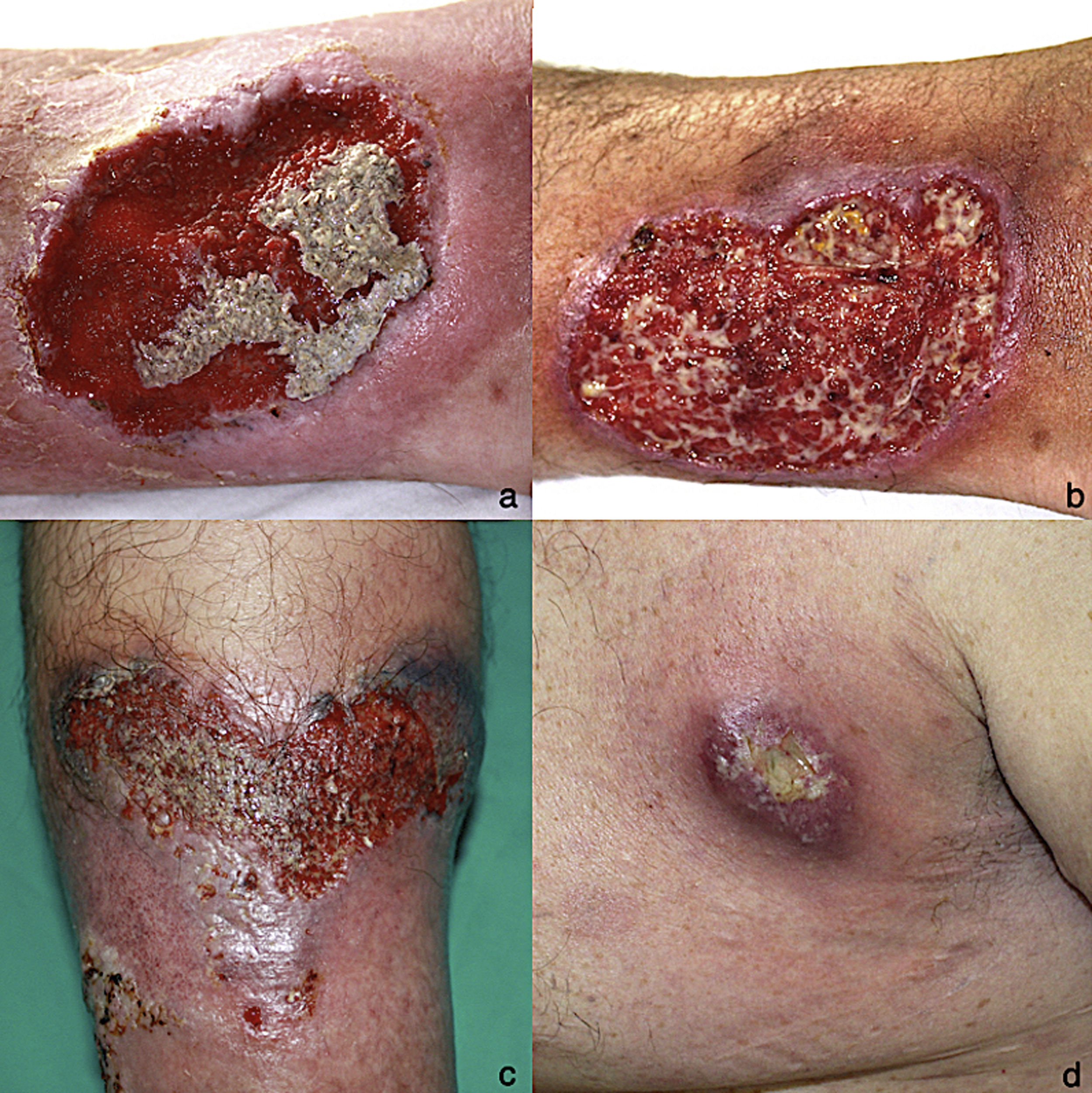
A, Ulcerative pyoderma gangrenosum on the right leg (April 2005). B, Ulcerative pyoderma gangrenosum on the right leg (October 2009). C, Detail of ulcerative pyoderma gangrenosum lesion (10 × 6 cm) on the left leg (July 2015). D, Ulcerative pyoderma gangrenosum in the left armpit (May 2017).
ND is a group of skin diseases characterized by the presence on histology of a predominantly neutrophilic inflammatory infiltrate, without evidence of infection. Each of these entities can be considered part of the same spectrum of diseases (and have overlapping clinical and histological characteristics), and can occasionally appear simultaneously. We have found only 13 published cases describing concomitant PG and SPD in a single patient, most of which were associated with monoclonal IgA gammopathy.2
There are 3 main factors that contribute to the pathogenesis of ND: abnormal expression of inflammatory molecules; altered neutrophil function; and genetic predisposition.1 Some studies have demonstrated overexpression of tumor necrosis factor alpha (TNF-α), interleukin 1 (IL-1) and its receptor, IL-17, and IL-8 in skin lesions in ND patients.1,3 TNF-α, IL-17, and IL-8 induce the activation and migration of neutrophils and the synthesis of matrix metalloproteinases MMP-2 and MMP-9, resulting in tissue damage.1,3
Up to one third of ND patients do not respond to first-line treatment (systemic corticosteroids combined with a corticosteroid-sparing agent). A recent semi-systematic review reported an overall response rate of 87% and a complete response rate of 67% in PG patients treated with TNF-α inhibitors, and found no significant differences between infliximab, adalimumab, and etanercept.4 Adalimumab is a therapeutic alternative for the treatment of PG associated with synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome (sometimes combined with low doses of methotrexate), pyoderma gangrenosum, acne, suppurative hidradenitis (PASH) syndrome,5,6 and PG refractory to conventional immunosuppressive therapies and other TNF-α inhibitors. Substitution of infliximab and etanercept with adalimumab has shown good results in the vast majority of patients,7 except for one who did not respond to either etanercept or adalimumab.8 In complex cases, intravenous immunoglobulins and ustekinumab have also been used with success (overall response rate: 66.7 and 66.6%, respectively).
We have found only 2 published cases of patients diagnosed with SPD who have been treated with adalimumab. De Encarnação Roque Diamantino et al9 reported a case in which the patient initially received topical and systemic corticosteroids, sulfone, sulfapyridine, and acitretin, without improvement. Complete lesion clearance was achieved after 6 weeks of treatment with adalimumab (40 mg every 2 wk) and a tapering dose of deflazacort. Versini et al10 described the case of a patient with SPD and MGUS who was refractory to multiple treatments, including infliximab and etanercept. Complete lesion clearance was observed after 5 months of adalimumab therapy (induction dose, 80 mg at wk 0 and 40 mg at wk 1; maintenance dose, 40 mg every 2 wk).
To our knowledge this is the first published report of a good response to adalimumab in a patient with concomitant PG and SPD. This drug is a potential therapeutic alternative for the treatment of ND refractory to other agents, and can provide rapid lesion improvement and control of recurrences.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: García del Pozo-Martín de Hijas MC, Agudo-Mena JL, Gómez-Sánchez ME, Escario-Travesedo E. Pioderma gangrenoso y dermatosis pustulosa subcórnea refractarios tratados con éxito mediante adalimumab. Actas Dermosifiliogr. 2020;111:887–889.


