


INTRODUCCION
Las queratodermias palmoplantares (QPP) constituyen un grupo muy heterogéneo de enfermedades caracterizado por un engrosamiento a nivel de las palmas y de las plantas como consecuencia de anomalías en la queratinización. La clasificación en función de las manifestaciones clínicas (difusa, focal, punctata) y los cambios histológicos es bastante compleja, pues suele existir solapamiento entre entidades. Se han descrito casos de asociación entre QPP y cáncer, en ocasiones con patrón de herencia autosómico dominante.
DESCRIPCION DEL CASO
Un varón de 43 años, cocinero de profesión, con antecedentes patológicos de hiperuricemia y dislipemia, en tratamiento con colchicina, consultó por lesiones en palmas y plantas, de aproximadamente 6-8 años de evolución, asintomáticas y de curso progresivo. Entre los antecedentes familiares, el paciente refería que su madre, fallecida por un cáncer de colon, había presentado el mismo problema sin que hubiera consultado ni recibido tratamiento por este motivo. El paciente no refería ninguna otra sintomatología sistémica, ni tampoco contacto o ingesta de componentes arsenicales.
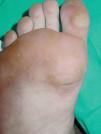
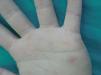
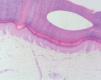
En la exploración física, en ambas palmas y plantas se apreciaban numerosas pápulas hiperqueratósicas redondeadas, poligonales, de tonalidad amarillenta-traslúcida (figs. 1 y 2). En el estudio histológico, con hematoxilina-eosina, se observó hiperqueratosis ortoqueratósica, acantosis y ausencia de laminilla cornoide (fig. 3), cambios compatibles con los de una queratodermia punctata palmoplantar. Se realizó determinación de marcadores tumorales (CEA, Ca19.9, β-HCG, α -FP, PSA, Cifra 21.1), que fueron normales y una radiografía de tórax donde no se encontraron hallazgos patológicos de interés. Se recomendó evitar los traumatismos locales en la medida de lo posible y tratamiento con calcipotriol tópico, que mejoró levemente las lesiones.
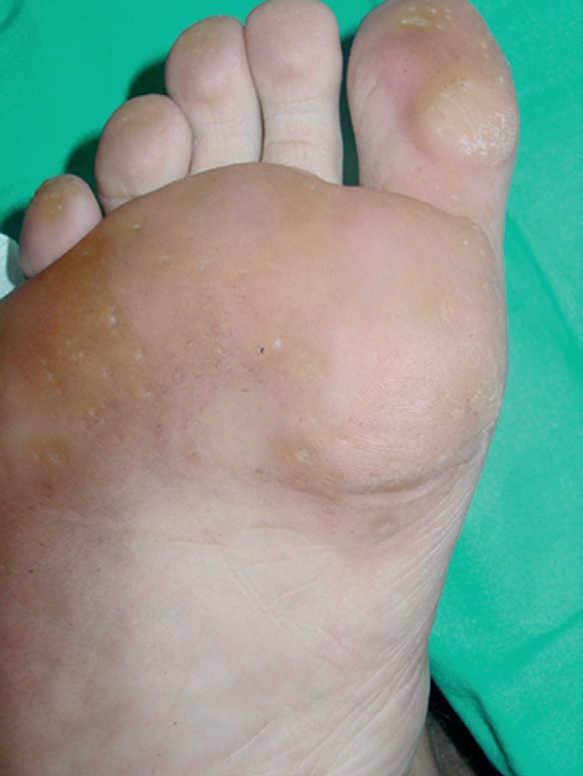
Fig. 1.--Lesiones hiperqueratósicas y deprimidas en la zona de la cabeza de los metatarsianos y de apoyo de los dedos de los pies.
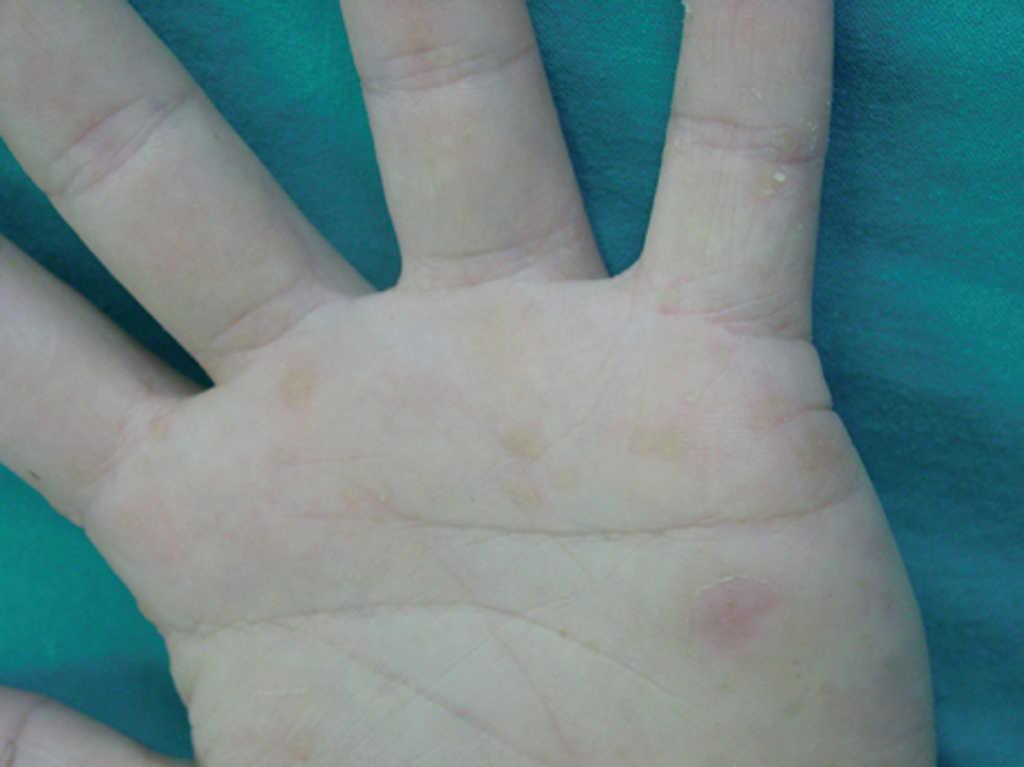
Fig. 2.--Lesiones papulosas amarillentas en las palmas de las manos.
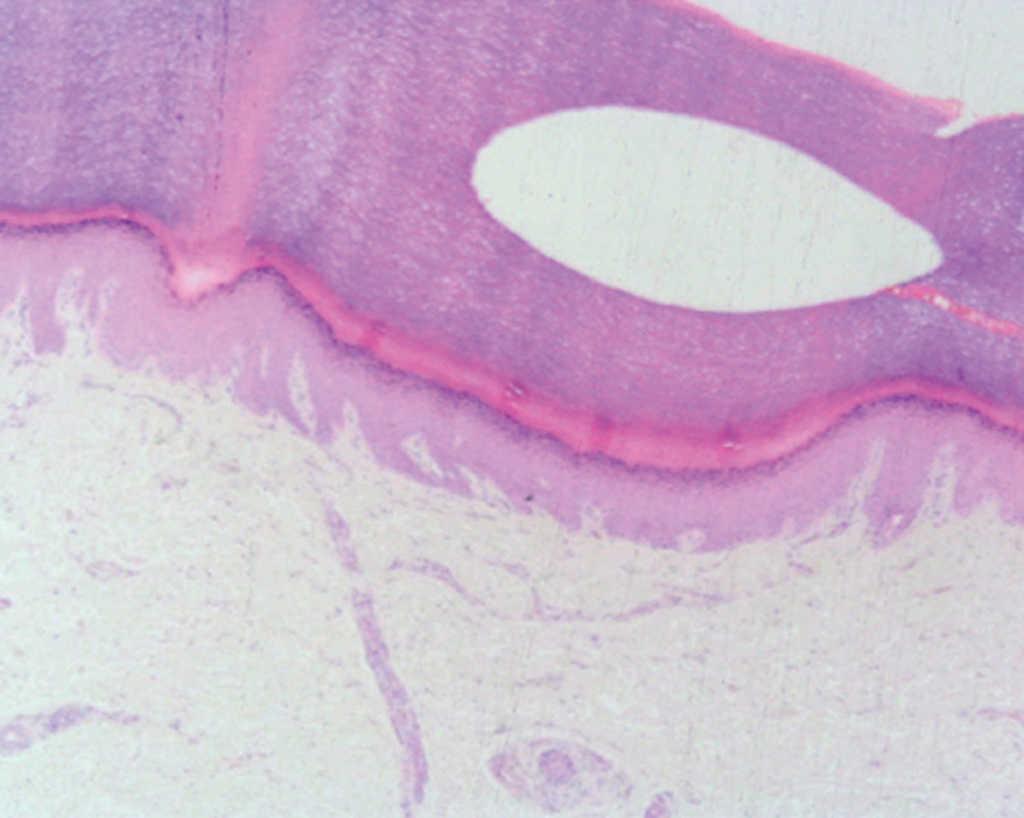
Fig. 3.--Acantosis, hiperqueratosis ortoqueratósica. (Hematoxilina-eosina, x10.)
DISCUSION
Existen cuatro tipos de QPP: difusa, focal, punctata y displasia ectodérmica palmoplantar. Asimismo, las QPP punctata pueden ser adquiridas (arsenicales, idiopáticas, poroqueratótica filiforme, paraneoplásicas, etc.) y hereditarias (tipo I: QPP de Buschke-Fisher-Brauer; tipo II: poroqueratosis punctata PP; tipo III: acroqueratoelastoidosis liquenoide). 1 La queratosis punctata de Buschke-Fisher-Brauer es una genodermatosis muy infrecuente, con una incidencia estimada de 1.17/100.000 2. Fue descrita por Buschke y Fischer en 1910 3 y Brauer en 1913 4. También se ha denominado keratoma disipatum hereditarium palmar y plantar de Brauer, queratoma diseminado, queratodermia maculosa diseminada simétrica palmoplantar de Buschke-Fischer, queratodermia punctata hereditaria y queratodermia palmoplantar papulosa, entre otras. El patrón de herencia es autosómico dominante, si bien aún no ha sido identificado el gen responsable, posiblemente debido al escaso número de familias afectadas recogido en la literatura médica 5.
Los trastornos en la queratinización que se producen en la QPP punctata, se podrían justificar por defectos en genes que intervienen en el desarrollo del epitelio y en la regulación de la expresión de la queratina. La asociación entre la QPP punctata y el desarrollo de los tumores se explicaría por una estrecha relación entre dos mutaciones, como puede ser una mutación en genes que regulan la producción de la queratina combinada con otra mutación en genes supresores de tumores 6.
La edad de inicio del proceso suele ser en la adolescencia o a partir de los 20 años, siendo muy rara su manifestación en niños. Se caracteriza por la presencia de múltiples lesiones queratósicas focales de 2-8 mm de diámetro, que comienzan como pápulas finas puntiformes, traslúcidas, las cuales evolucionan haciéndose opacas y verrucosas. En ocasiones, el centro queratósico central se elimina dejando una depresión central. En otros casos, las pápulas crecen y forman lesiones hiperqueratósicas de mayor tamaño que empeoran con el trabajo manual y pueden resultar dolorosas en las zonas de presión. No obstante, la mayoría de los pacientes están asintomáticos y el diagnóstico suele ser casual.
Las alteraciones que a continuación se enumeran se han visto asociadas con la queratodermia punctata palmoplantar: distrofia ungueal 7, malignidad 6, parálisis espástica, anodontia, artropatía HLA-B27, ceguera para los colores, ulcus duodenal y sindactilia 7. El tumor maligno que con mayor frecuencia se asocia es el adenocarcinoma de colon, si bien se han descrito pacientes que han desarrollado linfomas de Hodgkin y adenocarcinomas renales, de mama y pancreáticos 1,6.
El estudio histológico revela columnas compactas de hiperqueratosis masiva e hipergranulosis sobre un área bien delimitada, sin signos de inflamación en dermis y sin laminilla cornoide, dato que permite el diagnóstico diferencial con la poroqueratosis punctata palmoplantar. El diagnóstico diferencial incluye, además de la poroqueratosis punctata palmoplantar, las hiperqueratosis de roce, las verrugas virales, y la acroqueratoelastoidosis 5. La poroqueratosis punctata palmoplantar, también de herencia autosómica dominante y con manifestaciones clínicas superponibles, presenta paraqueratosis y laminilla cornoide en el estudio histológico, mientras que en la acroqueratoelastoidosis se suelen observar fibras elásticas desorganizadas 5.
En cuanto al tratamiento, se han utilizado queratolíticos, calcipotriol, retinoides tópicos y sistémicos, con resultados variables 5.
En el presente caso, la afectación idéntica en la madre del paciente, las características clínicas de las lesiones y los hallazgos histológicos, con ausencia de laminilla cornoide, han permitido realizar el diagnóstico del subtipo I de QPP hereditaria.
Declaración de conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Susana Mallo.
San Paulino, 18. 33204. Gijón. España.
susana2505@yahoo.com
Recibido el 11 de julio de 2005.
Aceptado el 21 de octubre de 2005.


