

The Aicardi–Goutières syndrome (AGS) – an inflammatory leukoencephalopathy with basal ganglia calcifications – is a type I interferonopathy (activation of the interferon [IFN] system), which leads to an increased expression of IFN-regulated genes.1 The phenotypic expression of AGS is heterogeneous and skin lesions, which may be key to achieve diagnosis.2
This is the case of a 19-year-old man from the Dominican Republic, with non-consanguineous parents, and an 18-year history of spastic paraparesis and delayed cognitive and motor development. After undergoing abdominal surgery in his country back in 2014, the patient experienced an isolated psychotic episode with mutism of unknown etiology.
In April 2020, he presented with chilblain-like acral lesions, which developed when he first came to Spain. Histopathological examination revealed the presence of necrosis and a dense perivascular lymphohistiocytic infiltrate with endothelial swelling and foci of thrombosis in dermal vessels. No dermal mucin deposition was observed. Direct immunofluorescence (DIF) test was not performed. The rest of the diagnostic work-up (including the ANA test and antiphospholipid antibodies test) was normal, and the patient was discharged with suspected chilblain-like lesions probably associated with asymptomatic SARS-CoV-2 infection.
In May 2021, after an upper respiratory tract infection, he was hospitalized for hallucinations, atypical behavior (paralysis, and occasional periods of mutism and perplexity with episodes of abnormal movements and maniform state), and a new outbreak of painful skin lesions. Among the additional tests performed this time, magnetic resonance imaging only revealed the presence of intracranial basal ganglia calcifications of linear distribution. The topography of lenticulostriate arteries revealed a bilateral and symmetrical distribution, without changes to the parenchyma or other associated findings, also seen on the brain CT scan. However, none of these findings helped clarify the etiology of pseudo-catatonia. CSF (cerebrospinal fluid) analysis was not performed.
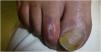
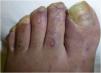
Dermoscopy revealed the presence of several, well-demarcated, depressed, porcelain-white 3–6mm plaques with a peripheral erythematous–violaceous halo on the dorsum of the patient's feet and toes (Figs. 1 and 2).


These lesions looked similar to the ones that have been described in Degos disease (DD), which is currently considered an interferonopathy.3 This, together with basal ganglia calcifications and the patient's past medical history of encephalopathy, was indicative of monogenic interferonopathy. A whole exome sequencing study was requested, and a homozygous pathogenic c.616C>T (p.His206Tyr) variant in the SAMHD1 gene was identified, supporting the diagnosis of AGS.
Classic AGS is a rare disease where nine genes involved in the intracellular nucleic acid metabolism have been identified with either recessive (mostly) or dominant traits, loss (LOF) or gain of function (GOF) gain pathogenic variants.2 In these pathways dysregulation triggers an abnormal IFN-1 response, which is responsible for neurological dysfunction and a broad range of inflammatory phenotypes in other AGS-like diseases (STING, ADA2, DNAE2, LSM11, RNU7-1, DNASE1L3, ACP5, POLA1, USP18, OAS1, CDC42, STAT2, ATAD3A).1
AGS can appear after several months of normal child development, with unspecific symptoms such as psychomotor delay and/or loss of acquired skills. This phase typically stabilizes after a few months. However, after some triggers, outbreaks of extra-neurological signs, mainly cutaneous, may occur. In fact, these are present in up to 50% of SAMHD1 related disease, which also associates common intracerebral vasculopathy.2
Chilblain-like cutaneous lesions have been described as painful lesions that exacerbate with cold and are often found in acral regions.4 They vary in severity from acrocyanosis and swelling or erythematous–violaceous plaques to the development of blisters, ulceration, and even severe digital necrosis. Although areas of atrophic skin have been reported,4 we have not found any description similar to DD.
Histopathological examination of chilblains in AGS patients has not drawn a uniform picture. A variable degree of thrombotic vasculopathy can be present and secondary ischemic signs such as epidermal necrosis, intraepidermal bullae formation, and degenerative changes like hyalinization have been reported.4
Psychiatric symptoms are exceptionally rare. According to Varesio et al.5 in their multi-center case series of 120 AGS participants, no patient presented psychiatric symptoms across the course of the disease. Currently, the association between AGS and catatonia or pseudo-catatonia, and the underlying mechanisms of psychiatric symptoms are still to be elucidated. We only found one case similar to ours reported by Ayrolles et al.,6 in 2020. They observed high levels of IFN-α in the CSF followed by a rapid normalization after clinical remission. Therefore, the exacerbation of psychiatric symptoms may be associated with certain triggers that may lead to high levels of IFN-α, although the disease was previously stabilized.
The role of IFN-I in blood vessel involvement has been shown after IFN-α treatment, with Raynaud phenomenon, cutaneous thrombosis, and ulceration.4 Furthermore, biopsies of DD lesions have demonstrated IFN-I signature in their pathophysiology.3
In conclusion, cold triggered Degos-like lesions can be a diagnostic key of a genetic interferonopathy. Hence, the role of the multidisciplinary team including a dermatologist in a patient with skin and psychiatric signs may be crucial for early diagnosis.
FundingThis study is funded by Instituto de Salud Carlos III (ISCIII) through the project FIS-PI21/01642, and co-funded by the European Union.
Conflict of interestThe authors declare that they have no conflict of interest.


