


Dystrophic epidermolysis bullosa (DEB) is a rare disease that represents a heavy burden for both the patient and the health care system. There are currently no data on the prevalence of DEB in Spain.
ObjectiveTo determine the prevalence of DEB in Spain.
MethodsWe used data from 3 incomplete population-based sources (hospital dermatology departments, diagnostic laboratories performing antigenic mapping, genetic testing or both, and the Spanish Association of Epidermolysis Bullosa Patients [DEBRA]) and combined them using the 3-source capture–recapture methodology.
ResultsWe identified 152 living DEB patients. The estimated prevalence of DEB was 6.0 cases per million (95% CI, 4.2–11.8) in adults and 15.3 (95% CI, 10.4–40.8) in children under 18 years of age. The data indicated that 77% of the patients were not being followed up in specialized centers of reference; 65% had not had a genetic diagnosis, and 76% were not members of DEBRA.
ConclusionsThe prevalence of DEB in Spain is 6.0 patients per million (95% CI, 4.2–11.8), a figure higher than previous estimates in many areas, but similar to those found in other southern Europe countries. The north–south difference may represent real geographic differences in prevalence, but it might be due to the fact that most of the data come from registries with a lower than expected catchment. Many patients are not being followed up in centers of reference, do not have genetic diagnosis, and are not members of patients’ associations, suggesting that there is room for considerable improvement in their care.
No existen datos sobre la prevalencia de la epidermólisis ampollosa distrófica en España (EAD). La EAD es una enfermedad rara que conlleva una gran carga para el paciente que la sufre y para el sistema de salud que le atiende.
ObjetivoDescribir la prevalencia de la EAD en España.
MétodosHemos empleado datos procedentes de 3 fuentes incompletas de pacientes: departamentos de Dermatología, 2 laboratorios de diagnóstico y la Asociación española de pacientes con epidermólisis ampollosa, DEBRA España, y los hemos combinado usando el método de captura-recaptura.
ResultadosHemos identificado 152 pacientes vivos. La prevalencia estimada de EAD fue de 6,0 casos por millón de habitantes (IC 95%: 4,2-11,8). La prevalencia en niños menores de 18 años fue de 15,3 por millón (IC 95%: 10,4-40,8). De acuerdo con el modelo de captura-recaptura el 77% no son seguidos en unidades de referencia, el 65% no tienen diagnóstico genético y el 76% no pertenecen a DEBRA.
ConclusionesLa prevalencia de EAD en España es de 6,0 pacientes por millón de habitantes (IC 95%: 4,2 a 11,8), un número mayor que el estimado en otras zonas del mundo, pero similar a otros encontrados en otros países del Sur de Europa. Este resultado puede ser debido a auténticas variaciones geográficas, o a que los otros registros recogen un número incompleto de casos. La mayoría de los pacientes no son seguidos en unidades de referencia, no tienen diagnóstico genético y no son miembros de la asociación de pacientes, lo cual quiere decir que su situación sociosanitaria es muy mejorable.
Epidermolysis bullosa (EB) is a group of inherited diseases in which the skin breaks and blisters easily following minor trauma. These disorders have been recently divided into 4 main groups depending on the ultrastructural level of skin cleavage at which the blister forms: EB simplex (EBS), junctional EB, dystrophic EB (DEB), and Kindler syndrome. Laboratory tests—electron microscopy (EM), antigenic mapping (AM), and/or molecular testing—are mandatory for diagnostic confirmation.1 DEB is a scarring form of EB of either autosomal recessive or dominant inheritance due to collagen VII gene mutations (COL7A1).2 The milder forms of recessive DEB (RDEB) and dominant DEB (DDEB) are clinically indistinguishable, and genetic testing is the only valid means of diagnosis. Patients with severe RDEB experience generalized severe mucocutaneous blistering and scarring accompanied by systemic manifestations that eventually lead to premature death.3 It has been estimated that the cumulative risk of death in severe DEB is 88.2% by age 45.4 DEB has a dramatic clinical and socioeconomic impact on both patients and their families, affecting personal, physical, emotional, and professional aspects of their life. Family burden questionnaire scores in DEB are similar to those observed in caregivers of cancer patients.5 In Spain, economic family burden is also significant because the public health system does not have centers of reference with multidisciplinary teams or outreach services and does not necessarily provide the basic necessities for these patients, such as non-adherent bandages and hand splints. The disease is also a burden on the public health system because complications inevitably arise since nearly all organs and body tissue are eventually involved.
Early intervention is critical because it will determine the quality of life and life expectancy of these patients. DEB patients also require information, specialized care, and social and financial support. The Spanish branch of the Dystrophic Epidermolysis Bullosa Research Association (DEBRA Spain) is very active and provides a great deal of support to affected families.
EB are considered rare diseases, meaning that their prevalence is less than 1 case per 2000 individuals and data on the prevalence of DEB in Spain is lacking. In rare diseases, accurate estimates of prevalence are important for planning health care and research but difficult to obtain. In Spain, patients with EB are mostly managed in centers with pediatric dermatology clinics. In August 2012, there were 28 patients with DEB on the Spanish National Registry of Rare Diseases (SNRRD)6 while DEBRA Spain had about 123 registered members, 70 of whom had been diagnosed by EM, AM, and/or genetic testing. The SNRRD and DEBRA Spain are the only registries of DEB patients in Spain and their data are probably incomplete.
The capture–recapture method was developed to merge data from different incomplete lists (e.g., incomplete registries) and to obtain an improved overall estimate of the total number of subjects, including those who are not on any of the lists, based on how much the different lists overlap.7 This method has been used in several medical fields8,9 to estimate the prevalence of skin diseases, such as rare ichthyosis,10 and is a very useful method for determining how complete registry data are.11 We used this technique to estimate the prevalence of DEB in Spain.
Patients and methodsThis was a prospective, population-based, cross-sectional study carried out using the capture–recapture technique. The study protocol was approved by the pertinent ethical committee (the Comité Etico de Investigación Clínica de Galicia – 2011/299) and by DEBRA Spain.
Lists of DEB patients were obtained from 3 sources: DEBRA Spain; dermatology departments in different areas of Spain that provide care to EB patients (Fig. 1); and 2 laboratories that perform advanced diagnostic tests, namely, the Hospital Clinic in Barcelona (AM) and CIEMAT-UC3M-CIBERER (AM and genetic testing of DEB). The specialists working in participating dermatology departments contacted dermatologists working in smaller public hospitals in their area to ask about known patients with DEB. Nearly all patients with DEB attend public hospitals because of the severity of the disease. All the participating centers provided a list of patients with DEB alive in October 2011, coded using the patient's initials and year of birth. Individual identifiers were matched after checking both initials and year of birth. We considered that patients were alive if there was evidence of contact with health providers in 2011 and no evidence of death in their records. In other cases, patients were contacted to confirm their life status. Doubts about matching were solved by contact between the data owners. For case definition we accepted clinical diagnosis by a dermatologist only if this had been confirmed either by means of EM, AM, or detection of COL7A1 mutations. Similarly, we only considered patients from DEBRA Spain whose diagnosis had been confirmed by EM, AM, or genetic testing. As not all patients had genetic testing to reliably distinguish between RDEB and DDEB, no distinction was made between these 2 forms of DEB.
 and 2 laboratories (purple dots).")
Analysis was done using 3-source capture–recapture analyses through log-linear modeling using the RECAP module in the Stata 10 (StataCorp L.P.) data analysis and statistical software package.12 This module produces several models to fit the data, and the best fit was selected on the basis of the lowest Akaike Information Criteria. The assumptions underpinning the capture–recapture method are explained in the discussion section.
As the denominator for prevalence, we used updated estimates from the last Spanish census (July 2011, Instituto Nacional de Estadística).13
ResultsWe individually identified 152 EB patients (64% had REB, 10% DEB and 26% were unclassified). Of these, 42 patients were present in 2 of the sources, and 17 in all 3. Eight doubts about matching were resolved after contact between the participating centers. A table showing the overlapping between the 3 data sources is available from the author. Fig. 2 shows the age distribution of these patients compared to that of the Spanish population as a whole.

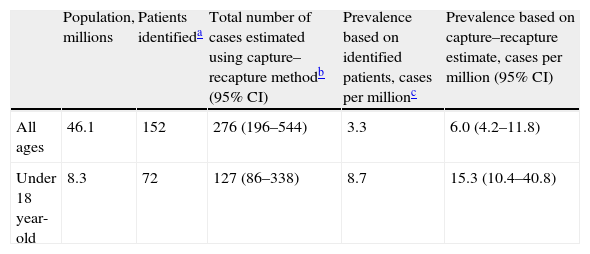
The capture–recapture method generates several models to fit the relationship between the different data sources. In our study, all models gave similar estimates, which is reassuring. The best-fitting log-linear model was a model with 2 pair interactions. Using this model, the best estimate of the non-captured population was 124 patients, which gives a total of 276 patients after the addition of the identified cases (Table 1). The resulting estimated prevalence of DEB is 6.0 cases per million (95% CI, 4.2–11.8). Of the patients in our lists, 58% were not under the care of centers of reference, 36% did not have a genetic diagnosis, and 56% were not members of the patient association. As the lists were incomplete, these percentages are higher when the results of the model are taken into account: 77% were not being followed by centers of reference, 65% did not have a genetic diagnosis, and 76% were not members of the patient association.
Prevalence of DEB in Spain.
| Population, millions | Patients identifieda | Total number of cases estimated using capture–recapture methodb (95% CI) | Prevalence based on identified patients, cases per millionc | Prevalence based on capture–recapture estimate, cases per million (95% CI) | |
| All ages | 46.1 | 152 | 276 (196–544) | 3.3 | 6.0 (4.2–11.8) |
| Under 18 year-old | 8.3 | 72 | 127 (86–338) | 8.7 | 15.3 (10.4–40.8) |
Assuming that data on children would be more accurate, we calculated the prevalence in children under 18 years of age. In this group, the best fitting model once again had 2 interaction terms. We detected 81 patients, and the model predicted 55 more undetected. The resulting prevalence was 15.3 cases per million (95% CI, 10.4–40.8) (Table 1).
DiscussionUsing the capture–recapture method we estimated the prevalence of DEB in Spain to be 6.0 cases per million in the overall population and 15.3 cases per million in children under 18 years of age. These estimated rates are higher than most previous prevalence reports. We have also shown that the data sources used were incomplete, a circumstance that justifies the use of the capture–recapture method to improve the accuracy of the estimate. However, even if we only take into account the actual patients identified without considering the predictions of the models, the minimum certain prevalence is 3.3 cases per million.
The validity of the capture–recapture method is based on the following 3 assumptions: a closed study population (e.g. not affected by migration); correct identification of subjects and the overlap between different data sources; and equal catchability (all patients must have the same probability of being captured by each source).
It is very likely that our study population was closed: DEBRA Spain, which maintains close contact with patients and sends information to their home addresses, was aware of only 1 patient leaving Spain over the last 10 years and none migrating to this country.
Correct identification of patients is certain. The initial matching of names and year of birth gave rise to 8 instances of doubt; all of these were subsequently resolved definitively when the data owners jointly reviewed their information.
The assumption that every patient in Spain has the same probability of being captured by each source is the most problematic of the 3 assumptions. If this criterion is not fulfilled it might mean that a hidden population exists that cannot be captured by any of the sources studied. This, in turn, would mean that our model underestimated prevalence rates. In the case of data from dermatology departments, there might be areas that were not adequately covered or hard-to-reach patients. To minimize this risk, we contacted hospitals all over the country and asked participants to collect data from other dermatology departments in their area. Due to the severity of DEB, patients are likely to seek frequent care in the public health system, minimizing the likelihood of a hidden population. With respect to the other 2 sources, the laboratory testing centers and DEBRA are clearly identified entities, well known to dermatologists and patients over Spain and accessible from anywhere in the country. Another factor likely to affect the probability of a patient being captured in the lists was age. The age distribution of the patients identified was markedly different from that of the general population (Fig. 2), with a preponderance of younger age groups. This is to be expected given that DEB is a disease associated with a significantly reduced life expectancy.4 However, part of this difference in age distribution could also be due to young patients requiring more health care and being more likely to have had genetic analysis and be members of DEBRA. This was confirmed by the higher overlap between sources in this age group. We believe that the assumptions of the capture–recapture model were adequately fulfilled in both age groups, but more strongly in the group of patients under 18, making it a more accurate estimate. If there are any errors in the estimates they are more likely to be due to the presence a hidden population, which would mean that our results are underestimated.
According to the results of our study, 77% of the patients are not being seen in centers of reference, 76% are not members of DEBRA Spain, and the majority of patients (65%) have not had an advanced laboratory diagnosis. This last finding may reflect the fact that genetic diagnosis has only been available in Spain since 2007.14 These results suggest that care for DEB patients could be much improved through the creation of an official network that would provide these patients with clinical care in centers of reference, routine non-molecular and molecular laboratory testing, social support through an association such as DEBRA, and overall institutional support.
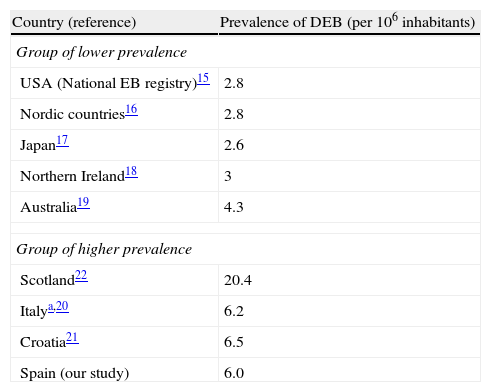
We found the estimated prevalence of DEB to be 6.0 cases per million inhabitants in Spain. Orphanet reports a prevalence of 7 cases per million; this figure was calculated as the mean of the highest and lowest values collected in the literature, a method that obscures geographical and methodological differences.15 Data from the US National EB Registry, currently the largest registry of EB patients and the most frequently quoted reference, indicate a prevalence of DEB of 2.8 cases per million.16 Other studies have found similar rates,17–20 except for some studies in Southern Europe21,22 and an extremely high prevalence of 20.4 per million reported in Scotland23 (Table 2). Such geographic variations in prevalence may be real or could be due to errors in the estimates. One likely error is that the population coverage of many registries (including the National EB Registry) may be lower than expected. Using the capture–recapture method to merge several data sources, as we did in our study, takes this problem into account and is likely to produce more accurate estimates. It is interesting to note a geographical trend and the fact that the higher prevalence figures tend to come from southern Europe, suggesting that the difference might be real. One factor that has been mentioned as the possible cause of high prevalence is the founder effect: higher prevalence in isolated countries or culturally closed communities might be associated with the presence of specific mutations in the population inherited from a common ancestor. Ethnic-specific recurrent mutations have been described in Spain as well as in other places and in certain ethnic groups. 14,24,25. In particular, the c.6527insC mutation, which arose around 3300 years ago and penetrated a bottleneck population about 1131 years ago,26 has a high prevalence among DEB patients from Spain14,25 and Chile,27 a country where a common Spanish predecessor is also plausible due to historical ties. Geographic differences in prevalence would have implications for health service planning and might make randomized clinical trials more feasible in countries with higher prevalence.
Reported prevalence of dystrophic epidermolysis bullosa in different areas.
Case definition is another area where errors may have occurred in previous studies and our own work. In the absence of confirmatory tests, some of the patients included may have been misdiagnosed, in particular those with non-Herlitz junctional EB and some types of scarring EB simplex. However, all of the patients in our study had EM, AM and/or genetic testing, and diagnosis of DEB can be considered certain. As most of the patients had not undergone molecular testing, the pattern of inheritance is unknown. Furthermore, since we did not take into account the severity of clinical manifestations, which can be vary considerably between severe RDEB, milder RDEB, and DDEB, it is difficult to evaluate the real burden of DEB on the Spanish health care system from our results. However, patients with more severe forms of RDEB are more likely to seek medical advice, undergo laboratory testing and join patient support associations. The findings in this respect are in line with the number of dominant versus recessive forms confirmed genetically (4 families and 70 patients respectively). Consequently, patients with more severe forms are more likely to be represented by our measure of prevalence.
In conclusion, the prevalence of DEB in Spain is 6.0 patients per million (95%CI, 4.7–11.8), a figure higher than previous estimates in other areas but in line with the presence of a recurrent mutation with a founder effect and similar to the rates found in other southern Europe countries. These data are useful for planning specialized care for these patients. Many patients are not being followed in specialized centers of reference, do not have genetic diagnosis and are not members of patient associations, suggesting that there is substantial room for improvement in their care. Capture–recapture methods should be used to assess the catchment of registries and to validate the accuracy of prevalence data from such registries.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors declare that they have no conflicts of interest.
We are grateful to DEBRA Spain, the patients and their families for their support and assistance to collect the required data. We are especially indebted to Esther Domínguez Pérez and Natividad Romero Haro (DEBRA nurses), and the following physicians: Virginia Fernández-Redondo (Complexo Hospitalario Universitario of Santiago), Ander Zulaica and José Luis Caeiro (Complexo Hospitalario Universitario of Vigo), Jesús del Pozo (Complexo Hospitalario Universitario of A Coruña), Mª Luisa Fernández y Mª Mercedes Lueiro (Hospital Lucus Augusti, Lugo), Cristina de las Heras (Hospital Arquitecto Marcide, Ferrol), María Luisa Zubiri (H. Miguel Servet of Zaragoza), José Francisco Carapeto (H.C.U. Lozano Blesa of Zaragoza), María Pilar Grasa. H.C.U. Lozano Blesa of Zaragoza, Yolanda Gilaberte (Hospital San Jorge of Huesca), Ana Morales-Callaghan (Hospital Ernest Lluch of Calatayud), Victoria Almeida (H. Txagorritxu of Vitoria), and Ana Tuneu (H. of Donostia).


