


A 30-year-old woman was seen for skin lesions on both legs that occurred in sporadic flares and were associated with localized itching. Notably, the patient's medical history included a diagnosis of Sjögren syndrome with sicca, based on compatible histological findings in a salivary gland biopsy and the presence of anti-Ro/SSa (Sjögren-syndrome-related antigen A) and anti-La/SSb+ (Sjögren-syndrome-related antigen β) antinuclear antibodies. The lesions consisted of nonpalpable purpura that persisted for approximately 5 days after exercise, prolonged standing, and stress, although the patient also reported the appearance of lesions in the absence of activity. In some cases the lesions were accompanied by pain and inflammation of the knees and ankles. The patient reported no fever or other systemic symptoms.
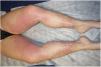
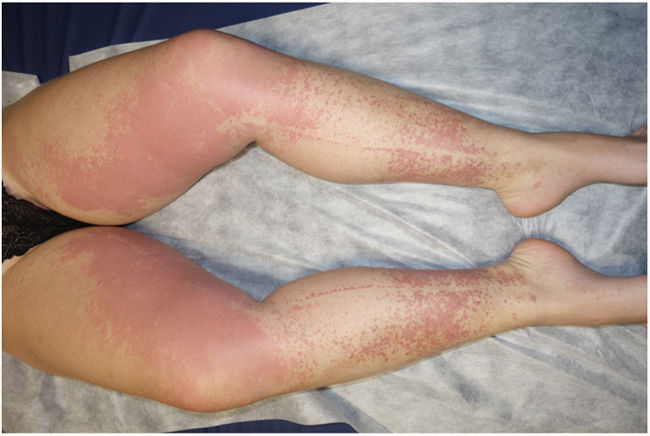
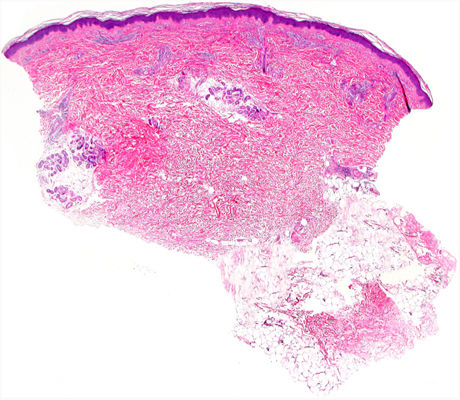
Examination revealed nonpalpable petechiae and purpuric spots, which did not blanch on finger pressure, distributed bilaterally and symmetrically on the legs, confluent lesions on the thighs, and isolated petechiae on the dorsal aspect of the feet (Fig. 1). A striking finding was the presence of the Rumpel-Leede phenomenon on the insides of both legs, coinciding with the seam of the pants.
In addition to the aforementioned anti-Ro/SSa and anti-La/SSb antinuclear antibodies, laboratory analyses revealed the following: elevated erythrocyte sedimentation rate (ESR; 76mm); rheumatoid factor, 84IU/mL; IgG, 2680mg/dL (polyclonal hypergammaglobulinemia on immunofixation). The remaining tests (complete blood count, coagulation study, basic biochemistry, serology, and complement analysis) revealed no alterations.
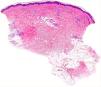
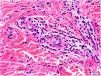
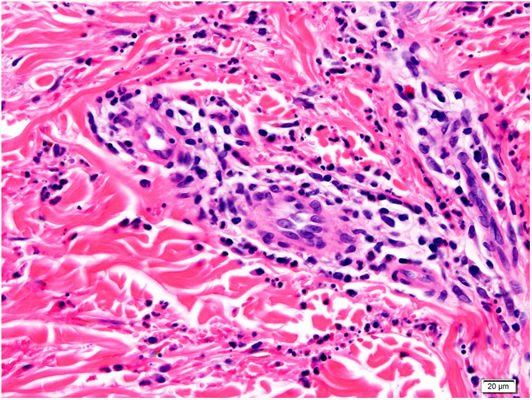
Histology of a biopsy of one of the purpuric macules showed a dense, lymphocytic and polymorphonuclear perivascular infiltrate with leukocytoclasia, without fibrinoid necrosis, in the upper papillary and reticular dermis (Figs. 2 and 3). Direct immunofluorescence (DIF) was negative.
What Is Your Diagnosis?
DiagnosisHypergammaglobulinemic purpura of Waldenström.
CommentA diagnosis of hypergammaglobulinemic purpura of Waldenström was established based on the recurrent flares of purpura on the lower limbs, the patient's clinical history (Sjögren syndrome), and the laboratory (hypergammaglobulinemia and elevated rheumatoid factor and ESR levels) and histological findings.
Hypergammaglobulinemic purpura of Waldenström is a rare entity that is common in middle-aged women and consists of flares of petechial lesions. These lesions predominantly affect the lower limbs, can coalesce to form purpuric spots (as seen in our patient), and rarely become palpable. Lesions can be accompanied by sensations of itching, burning, or even pain. Flares occur at highly variable intervals, ranging from weeks to years. In some cases the clinical picture can be accompanied by lymphadenopathy, arthritis, or fever. Relapses are usually triggered by increased hydrostatic pressure (e.g. due to prolonged standing).1
Two forms of purpura of Waldenström are recognized: primary, which is not associated with other diseases; and secondary, which is more frequent, and is associated with autoimmune disorders, granulomatous diseases, and blood dyscrasias. The secondary form is most commonly associated with Sjögren syndrome,2 followed by lupus erythematosus,3 Raynaud phenomenon, and multiple myeloma.
Key laboratory findings include polyclonal hypergammaglobulinemia, due to increased levels of IgG, as well as IgA and IgM.4 Elevated ESR levels typically coincide with flares. Rheumatoid factor is usually present at high titers. Other potential findings include usually mild alterations in the complete blood count (anemia, leukopenia), positive ANA titers, and, less frequently, hypocomplementemia or cryoglobulinemia, which can cause diagnostic confusion with other autoimmune diseases.
Histology reveals 2 equally frequent patterns: leukocytoclastic vasculitis and lymphocytic perivascular infiltrate without leukocytoclasia. The infiltrate is usually predominantly polymorphonuclear in early lesions (less than 24h after onset), with hematic extravasation and fibrinoid necrosis, later progressing to a mononuclear or mixed infiltrate. DIF findings (IgG, IgA, and IgM) are rarely positive, and knowledge of time since lesion onset is essential for interpretation of this parameter.
The differential diagnosis includes leukocytoclastic vasculitis and urticaria vasculitis. The latter was considered less likely in our case: the lesions were highly purpuric, slightly pruritic, and painless, without residual hyperpigmentation; hypocomplementemia was absent; and DIF findings were negative. Moreover, the presence of hypergammaglobulinemia further supported a diagnosis of purpura of Waldenström.
The benign and generally asymptomatic nature of this disease makes use of aggressive treatments unnecessary in most cases. Relative rest with legs raised is usually recommended, occasionally accompanied by medium-potency topical corticosteroid treatment. Cases treated with hydroxychloroquine, indomethacin, plasmapheresis, aspirin, colchicine, and maintenance oral corticosteroids have also been described, with variable results.
Conflicts of InterestThe authors declare that they have no conflicts of interest.


