














INTRODUCCION
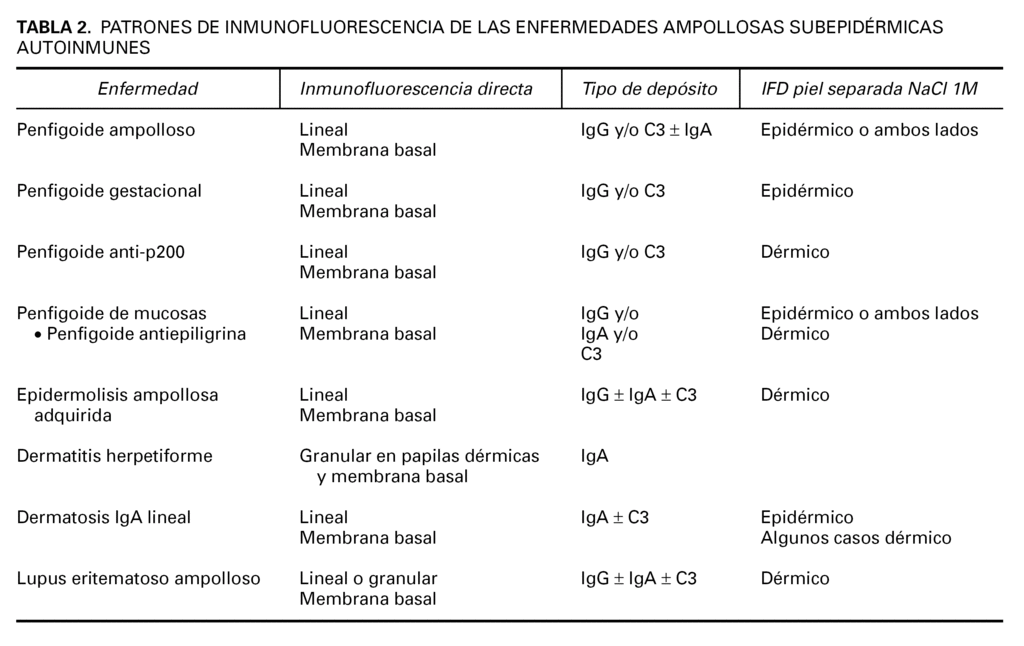
Las enfermedades ampollosas subepidérmicas autoinmunes
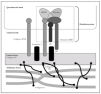
La unión dermoepidérmica es una estructura de alta complejidad molecular formada por los queratinocitos basales, la membrana basal epidérmica y la parte superior de las papilas dérmicas (fig. 1). El ataque autoinmune contra la unión dermoepidérmica desencadena un grupo de enfermedades denominadas enfermedades ampollosas subepidérmicas autoinmunes (EASA) 1,2. Las EASA tienen un mecanismo patogénico común por el que autoanticuerpos contra determinadas moléculas de la unión dermoepidérmica inducen una reacción inflamatoria en el área de las estructuras diana y una pérdida de función de moléculas de adhesión, con la subsiguiente formación de ampollas subepidérmicas y lesiones secundarias 3.
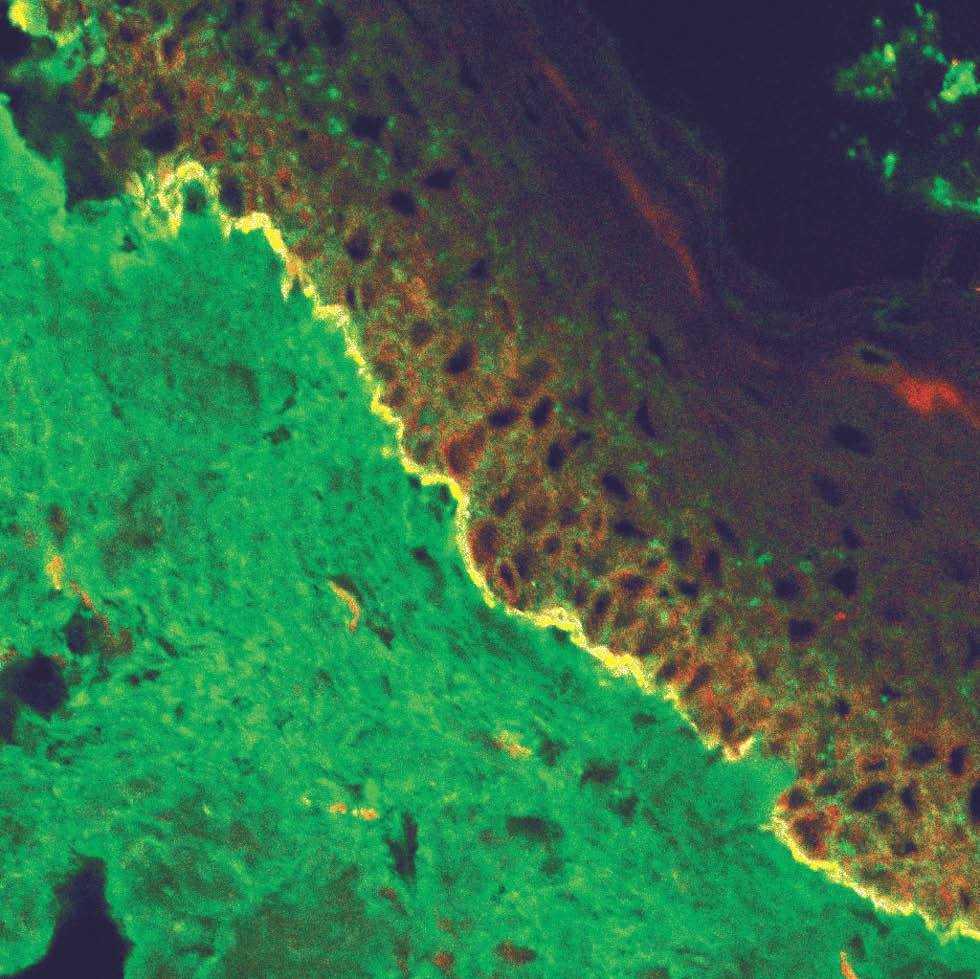
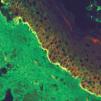
Fig. 1.--Imagen de microscopía confocal de la unión dermoepi-dérmica.
La EASA más frecuente es el penfigoide ampolloso, pero también pertenecen al grupo el penfigoide gestacional, penfigoide de mucosas, penfigoide anti-p200, liquen plano penfigoide, epidermolisis ampollosa adquirida, dermatosis IgA lineal, lupus eritematoso ampolloso y dermatitis herpetiforme (tabla 1). Dentro de las EASA es característico el solapamiento clínico e histopatológico entre distintas entidades.

Los estudios inmunológicos, que nacieron para la investigación y de hecho han definido nuevos subtipos de EASA, se han ido incorporando progresivamente a la clínica, y hoy en día resultan absolutamente imprescindibles para el diagnóstico de este grupo de enfermedades.
A pesar de todo, los estudios inmunológicos no han hecho más que incrementar la sensación de inmenso desconocimiento que se tiene al profundizar en las EASA. En la práctica ocurre que casos clínicamente sospechosos de un tipo de EASA se corresponden inmunológicamente con otra 4,5. Hay superposiciones clínico-inmunológicas y exclusivamente inmunológicas. Un ejemplo de esto último es una serie de casos de la literatura con clínica de penfigoide ampolloso y clara reactividad frente a colágeno XVII y desmogleína 3 sin las lesiones en mucosas características del pénfigo vulgar 6-13.
Las superposiciones se atribuyen en muchos casos a la expansión de epítopos, un proceso por el cual, debido al daño tisular por un proceso antiinflamatorio autoinmune, la exposición de un antígeno previamente «secuestrado» conduce a una respuesta autoinmune secundaria frente a este antígeno 14.
Los métodos diagnósticos en las EASA tienen 3 objetivos:
1. Distinguir las EASA de otros grupos de enfermedades vesiculoampollosas:
a) Enfermedades ampollosas hereditarias.
b) Enfermedades infecciosas (virales y bacterianas).
c) Otras enfermedades de mecanismo inmunológico (por ejemplo eritema exudativo multiforme, reacciones medicamentosas).
d) Miscelánea: porfiria cutánea tarda, bullosis diabeticorum, ampollas de origen físico y eczema dishidrótico.
2. Distinguir las EASA entre sí.
3. En casos concretos monitorizar el curso de la enfermedad, detectar precozmente los brotes y ajustar el tratamiento.
Estructura y proteínas principales de la unión dermoepidérmica
La unión dermoepidérmica es una estructura altamente especializada que actúa como vehículo de comunicación entre epidermis y dermis. Hoy no se mantiene la visión estática, puramente física de la unión dermoepidérmica, sino que se considera que realiza las siguientes funciones:
1. Adhesiva: sustrato para la adhesión de los queratinocitos basales, anclaje dermoepidérmico, transmisión mecánica entre epidermis y fibras elásticas y colágenas dérmicas.
2. Barrera: regulación de la permeabilidad de macromoléculas en ambos sentidos.
3. Señalización celular: transmisión de señales de diferenciación, morfogénesis y apoptosis epidérmica, permeabilidad de células inflamatorias y molde para la reparación tisular.
Como se ha dicho anteriormente, la unión dermoepidérmica consta de la membrana basal y de estructuras diferenciadas de los queratinocitos basales y la dermis papilar 15 (fig. 2).
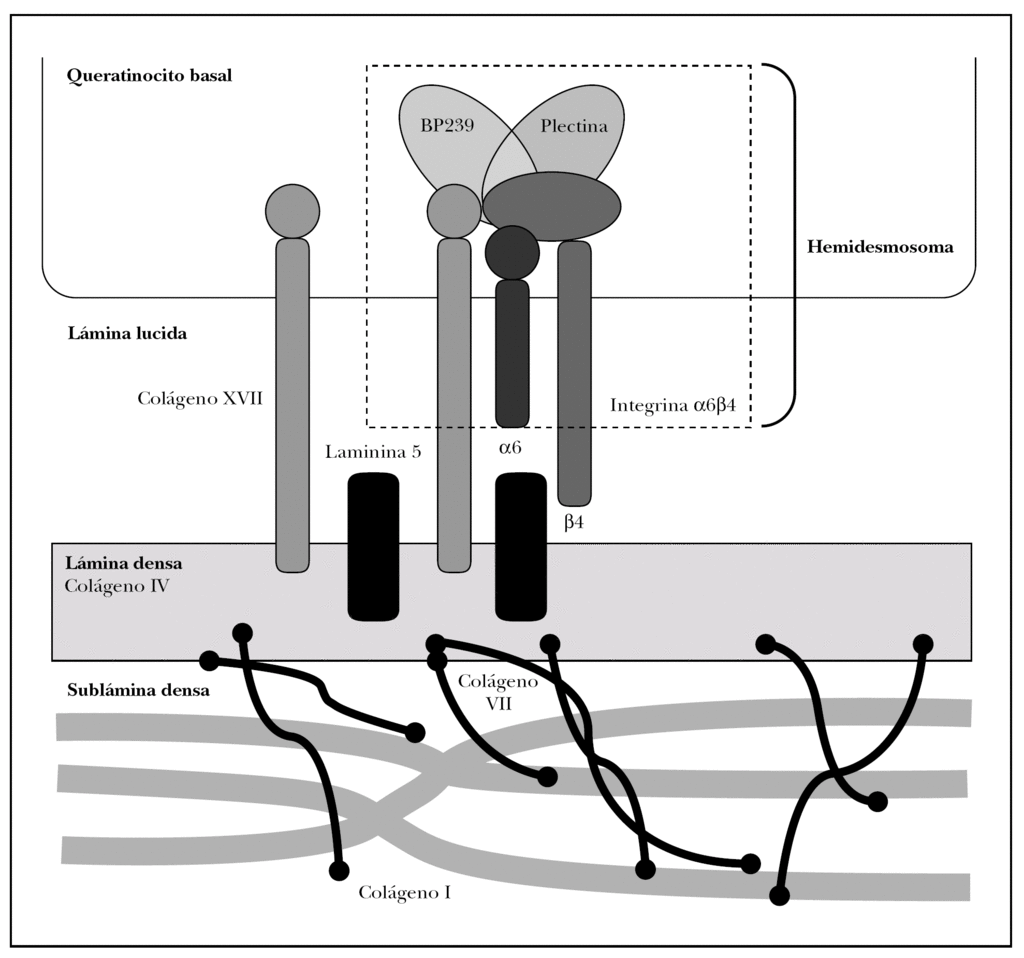
Fig. 2.--Estructuras y proteínas principales de la unión dermoepidérmica.
Los queratinocitos basales presentan un citoesqueleto con filamentos intermedios de queratina 5 y 14 que se conectan con unas estructuras basales llamadas hemidesmosomas y una membrana plasmática basal especializada.
La membrana basal epidérmica se puede subdividir a su vez en varias zonas, caracterizadas inicialmente por microscopía electrónica por su densidad a los electrones. En dirección epidermis-dermis hay una zona clara llamada lámina lúcida y una oscura llamada lámina densa. Hoy se sabe que la lámina lúcida es un espacio artefactual creado por deshidratación durante el procesamiento convencional de tejidos, pero en la presente revisión preferimos mantener este término por su estabilidad en la literatura 16.
La interfase entre la lámina densa y la dermis papilar se denomina sublámina densa, y contiene estructuras denominadas fibrillas de anclaje y placas de anclaje junto a proteínas fibrilares dérmicas.
Las moléculas conocidas de interés para el estudio de la patología autoinmune se localizan en los queratinocitos basales, la membrana basal y las fibrillas de anclaje. Describimos a continuación tan sólo las principales moléculas implicadas en patología autoinmune o en sus técnicas diagnósticas.
Queratinocito basal y lámina lúcida
Se tratan conjuntamente por ocupar los hemidesmosomas ambas regiones. Los hemidesmosomas son estructuras con función de anclaje y señalización celular que están situados en la membrana plasmática basal del queratinocito basal. Las proteínas fundamentales del hemidesmosoma son colágeno XVII, plectina, BP230 e integrina α6β4.
Colágeno XVII. Molécula también conocida como BPAG2 (bullous pemphigoid antigen 2) o BP180 (bullous pemphigoid 180 KDa) 2,15. Es la proteína implicada con más frecuencia en la patología autoinmune de la unión dermoepidérmica y la mejor conocida 17,18.
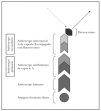
Es una glucoproteína transmembrana (fig. 3). Se ha clasificado dentro de la familia de los colágenos como colágeno XVII (COL17A1) porque en su extremo C-terminal existen 15 secuencias repetidas tipo colágeno (Gly-X-Y). Su extremo N-terminal es intracelular y se une a BP230, plectina y a la subunidad b4 de la integrina α6β4. El extremo C-terminal se extiende a través de la lámina lúcida y llega a la lámina densa 19.

Fig. 3.--Estructura del colágeno XVII o BP180.
El colágeno tipo XVII existe in vivo en dos formas, la completa, de 180 kDa, y el dominio extracelular de 120 kDa que se separa por proteolisis in vivo. Este ectodominio se llama LAD-1. Los dominios extracelulares se ensamblan de 3 en 3 formando triples hélices tipo colágeno 20.
Es particularmente importante en la patología cutánea el segmento extracelular del colágeno XVII, más próximo a la membrana plasmática, llamado NC16A (non-collagenous 16), que es el decimosexto segmento no colágeno. Se une a la subunidad a6 de la integrina α6β 4 21.
Se han demostrado anticuerpos séricos contra el colágeno XVII en el penfigoide ampolloso, penfigoide gestacional, penfigoide de mucosas, dermatosis IgA lineal y liquen plano penfigoide. En penfigoide ampolloso, penfigoide gestacional y dermatosis IgA lineal puede detectarse inmunidad contra NC16A. En algunos penfigoides de mucosas hay reacción contra epítopos más C-terminales. El fragmento LAD-1 está implicado en dermatosis IgA lineal y penfigoide ampolloso.
Se ha demostrado patogenicidad de anticuerpos contra colágeno XVII en un modelo de ratón 22. En la epidermolisis ampollosa juntural no Herlitz se encuentra mutado el gen del colágeno XVII.
BP230. Es una proteína de 230 kDa también denominada BPAG1 (bullous pemphigoid antigen 1) 2,15. Se trata de una proteína intracelular del queratinocito basal situada en el complejo hemidesmosómico. Pertenece a la familia de las plaquinas. Su función es unir los filamentos intermedios del citoesqueleto al hemidesmosoma. Su extremo C-terminal se asocia a los filamentos intermedios de queratina. Su extremo N-terminal se une al colágeno XVII y subunidad b 4 de la integrina α6β 4, entre otras.
En penfigoide ampolloso y dermatosis IgA lineal aparecen autoanticuerpos contra BP230, pero se duda de que sean patogénicos; más bien se cree que su producción se induce secundariamente a la reacción inflamatoria en la unión dermoepidérmica.
No hay mutaciones conocidas de BP230 en la patología ampollosa hereditaria.
Plectina. La plectina es una proteína globular situada en el lado citoplasmático del hemidesmosoma, también de la familia de las plaquinas. Se une por su extremo C-terminal a los filamentos intermedios de queratina y por su extremo N-terminal a la subunidad b4 de la integrina α6β4, BP230 y actina23.
Se han descrito algunos casos de reactividad contra plectina en patología autoinmune, sobre todo penfigoides ampollosos, pero tiene una importancia secundaria 24-26.
La plectina está mutada en un tipo de epidermolisis ampollosa simple (epidermolisis ampollosa simple con distrofia muscular de cinturas).
Integrina α6β4. Las integrinas son glucoproteínas transmembrana que median la adhesión célula-matriz o célula-célula con capacidad de transducir señales reguladoras de expresión génica y ciclo celular. Son heterodímeros, lo que significa que consisten en 2 subunidades distintas entre sí 2,15. Los queratinocitos basales expresan a2b1 (membranas laterales y apical), α3β1 (en membranas laterales y en membrana plasmática basal, pero no asociada al hemidesmosoma, se une a laminina 5) y α6β4 (proteína hemidesmosómica).
La subunidad β4 tiene un extremo C-terminal intracelular y un extremo N-terminal extracelular. El dominio C-terminal se une a plectina y colágeno XVII y el dominio extracelular se une a la laminina 5; por tanto, media la conexión del hemidesmosoma por un lado, con los filamentos intermedios de queratina, y por otro lado con lámina densa.
La subunidad α6 se une al dominio NC16A del colágeno XVII.
Las dos subunidades son antígenos en el penfigoide de mucosas oral y ocular. También existe un modelo animal. La mutación de cualquiera de las 2 subunidades conduce a la epidermolisis ampollosa juntural con atresia pilórica.
Lámina densa
La lámina densa se compone principalmente de colágeno IV, lamininas 5, 6 y 10 y proteoglicanos.
Colágeno IV. Es un heterotrímero formado a partir de 6 genes diferentes, de los cuales en la piel se expresan COL4A1 (subunidad a1[IV]), COL4A2 (subunidad α2[IV]), COL4A6 (subunidad α6[IV]), COL4A5 (subunidad α5[IV]) 2. En común con otros colágenos tiene un largo dominio triple hélice tipo colágeno; posee 2 dominios no colágenos globulares en los extremos C-terminal (NC1) y N-terminal (NC2) que permiten el ensamblaje con la propiedad de crear redes colágenas planas.
El dominio NC1 de α3(IV) es la diana en el síndrome renopulmonar de Goodpasture. En un paciente con insuficiencia renal y enfermedad ampollosa subepidérmica se detectó IgG contra el dominio NC1 de α5(IV).
El colágeno IV es un importante marcador de la lámina densa en los métodos diagnósticos.
Laminina 5. Las lamininas son una familia de glucoproteínas heterotriméricas que cumplen funciones estructurales y de señalización. Cada laminina está formada por tres subunidades (α,β,γ) 2,15. La laminina 5 (α3, β3, γ2) se encuentra en la interfase entre lámina lúcida y lámina densa e interacciona con las integrinas del hemidesmosoma y la membrana plasmática basal del queratinocito.
La laminina 5 se conoce también como epiligrina, y un tipo de penfigoide de mucosas con autoinmunidad contra laminina 5 se denominaba penfigoide antiepiligrina 27-29. La mayoría de los pacientes con penfigoide de mucosas y autoinmunidad contra laminina 5 reconocen la subunidad α3 30.
Sublámina densa
Las fibrillas de anclaje son unas estructuras arciformes observables mediante microscopía electrónica, que se componen fundamentalmente de colágeno VII 31.
Colágeno VII. Es otra proteína de la familia del colágeno; es un homotrímero formado por tres cadenas idénticas de 290 kDa. Tiene un dominio globular no colágeno N-terminal (NC1) donde se localizan los epítopos inmunodominantes. En el extremo C-terminal se sitúa el dominio NC2 2,15,32-34.
Es el antígeno implicado en la epidermolisis ampollosa adquirida, el lupus eritematoso sistémico ampolloso y algunos casos de dermatosis IgA lineal 35-37.
Su alteración de causa genética se traduce en distintos tipos de epidermolisis ampollosa distrófica.
MÉTODOS DIAGNOSTICOS
Dividimos los métodos diagnósticos en dos grandes grupos: aquellos que detectan autoanticuerpos en tejidos o suero y el resto de métodos.
Métodos diagnósticos sin detección de autoanticuerpos
Histopatología convencional
La biopsia convencional fijada en formol es un punto de partida imprescindible en el diagnóstico histopatológico 2,38. La elección del lugar de biopsia es muy importante (fig. 4). Debe estudiarse el borde de la ampolla o vesícula para observar el lugar de despegamiento bajo el microscopio. La presencia de un infiltrado rico en eosinófilos, neutrófilos o pobre en células inflamatorias es orientativa pero nunca diagnóstica (fig. 5).
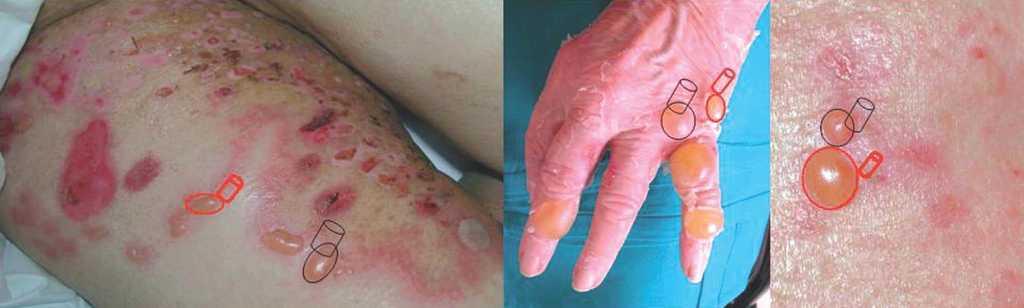
Fig. 4.--Lugares recomendados para la realización de biopsias para estudio convencional (negro) y para inmunofluorescencia (rojo).
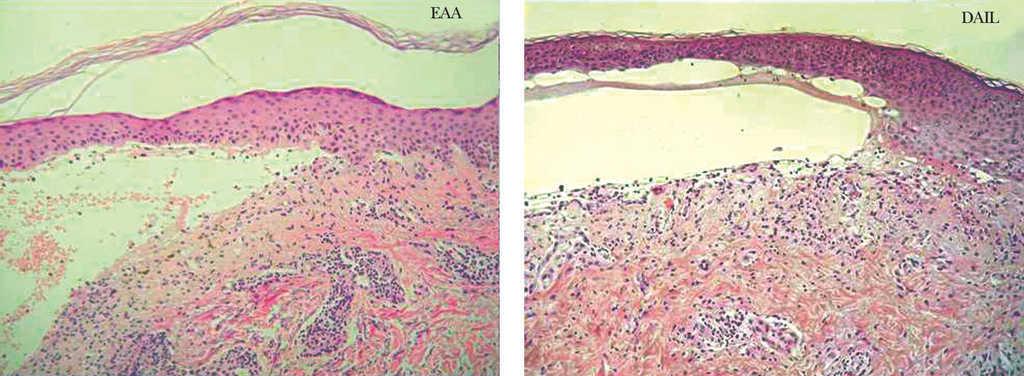
Fig. 5.--Tinción con hematoxilina-eosina de una epidermolisis ampollosa adquirida (EAA) y una dermatosis IgA lineal (DAIL). En ambas se observa un despegamiento subepidérmico y un infiltrado inflamatorio moderado con neutrófilos. La histopatología convencional no distingue unas enfermedades ampollosas subepidérmicas de otras.
Mapeado de antígenos mediante inmunohistoquímica
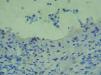
Para localizar el nivel de separación dentro de la unión dermoepidérmica es posible realizar una tinción de alguna proteína conocida mediante inmunohistoquímica y visualizar la situación de ésta con respecto al despegamiento patológico. La ventaja es que puede realizarse en muestras ya fijadas en formol e incluidas en parafina. Se han marcado las queratinas 5 y 14 del queratinocito basal, las lamininas y el colágeno tipo IV de la lámina densa. El colágeno IV es el más utilizado (fig. 6) 2.
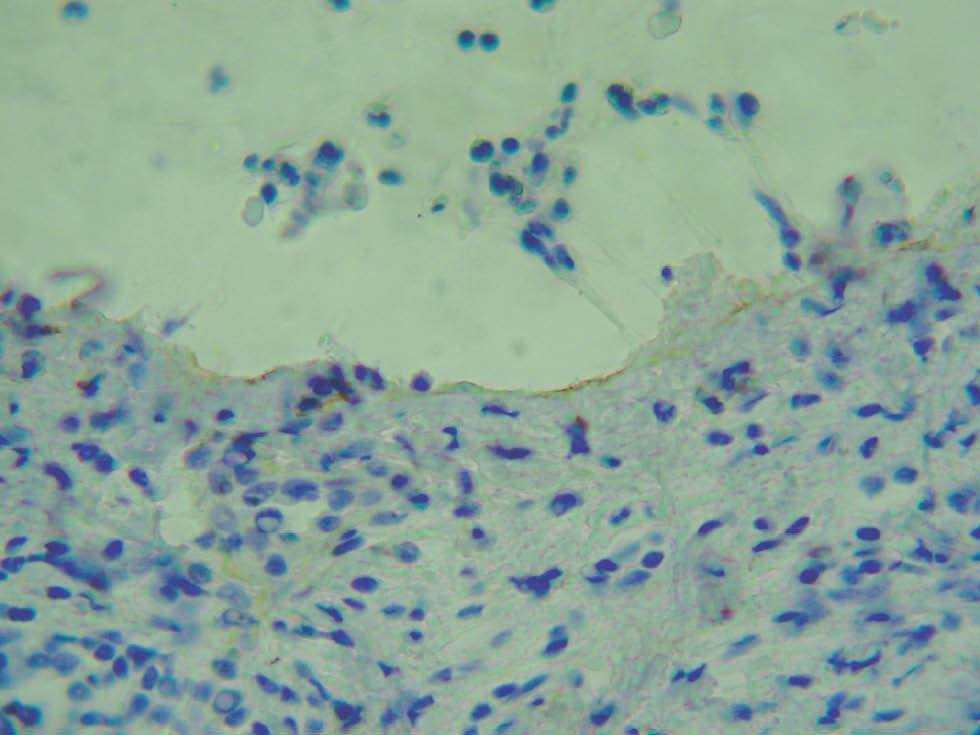
Fig. 6.--Tinción inmunohistoquímica para colágeno IV, que se observa de coloración parda en el suelo del despegamiento patológico en un caso de penfigoide ampolloso.
Microscopía electrónica convencional
Es una técnica lenta y complicada que no aporta ventajas en el diagnóstico de las EASA al no dar información sobre los depósitos autoinmunes. En trabajos de investigación antiguos se caracterizaron las distintas EASA mediante microscopía electrónica con localización fina del despegamiento y descripción ultraestructural de cambios celulares, pero en la actualidad no es un campo muy activo de investigación 38.
Métodos diagnósticos con detección de autoanticuerpos
El objetivo de estos métodos es detectar la presencia de autoanticuerpos o complemento depositados en tejidos o circulantes en suero y caracterizarlos en clase y especificidad antigénica.
Detección de depósitos autoinmunes en piel
Inmunofluorescencia directa
Su objetivo es detectar depósitos de inmunoglobulinas o complemento en la unión dermoepidérmica 2,38-42.
IFD convencional
1. Toma de la biopsia. La elección del lugar de la biopsia para inmunofluorescencia directa es clave para la rentabilidad diagnóstica 40,43. Nunca deben biopsiarse áreas de ampolla, ya que los depósitos autoinmunes se encuentran distorsionados por la separación dermoepidérmica y pueden haber sido degradados por la respuesta inflamatoria, dando lugar a falsos negativos. En la familia del penfigoide ampolloso y en la epidermolisis ampollosa adquirida son rentables las áreas adyacentes a las ampollas. Tradicionalmente se ha recomendado la realización de una cuña cutánea radial que incluyera zona de despegamiento y zona periférica, la división de la cuña con el bisturí y el envío de la zona periférica para estudio de inmunofluorescencia directa. El punch o sacabocados ha sido desaconsejado por algunos autores por la posibilidad de separar la epidermis con el movimiento rotacional. En nuestro medio su amplísima difusión y la familiaridad de los patólogos con este formato estándar de biopsia hace que, a pesar de todo, resulte muy práctica la toma cuidadosa de una muestra independiente para inmunofluorescencia con sacabocados, que es diagnóstica con un tamaño tan pequeño como 3 mm de diámetro (fig. 4). En nuestra experiencia nunca se han producido falsos negativos por la toma de biopsias de inmunofluorescencia con sacabocados.
El área corporal puede influir en la rentabilidad de la biopsia. En un estudio se vio que la biopsia de las piernas es menos rentable en el penfigoide ampolloso 44.
En el penfigoide de mucosas hay que elegir las áreas menos traumáticas para biopsiar. Si hay lesiones en piel deben ser éstas las que se biopsien. Si hay lesiones orales y oculares deben elegirse las primeras. Si sólo hay lesiones oculares hay que valorar el beneficio-riesgo de la realización de la biopsia, dado que ésta puede inducir inflamación y cambios cicatriciales.
2. Transporte y conservación de la muestra. En los centros que disponen de un laboratorio de Anatomía Patológica con unidad de inmunofluorescencia puede transportarse la biopsia en fresco en una gasa empapada en suero isotónico para realizar congelación inmediata. En caso de que la biopsia se haga fuera del horario de recogida de muestras del laboratorio, y vaya a ser procesada al día siguiente lo óptimo es conservarla en suero isotónico a temperatura ambiente. Se ha demostrado que esto no sólo no altera la muestra, sino que además limpia la fluorescencia residual por extravasación en dermis y depósito inespecífico 45,46.
En el ámbito anglosajón es muy utilizado el medio de Michel. Se ha comprobado que las biopsias conservadas en este medio resisten hasta un año a temperatura ambiente. En España no es fácil conseguirlo de forma comercial 47-50.
Hay comunicaciones aisladas de buena conservación de biopsias en medios como la miel 51.
3. Procesamiento y tinción. Hay varios métodos de procesamiento de la biopsia. En el más utilizado la muestra se congela utilizando nitrógeno líquido y un medio comercial de congelación tisular. Se utiliza un protocolo mediante el cual el tejido se congela lentamente y así se evitan daños y artefactos. La muestra puede guardarse durante períodos prolongados a 80 °C o proceder al corte y tinción.
El criostato es un aparato diseñado para cortar muestras en congelación. Suelen generarse criosecciones de 4 a 6 micras, aunque se ha demostrado que realizar secciones aún más finas, menores de 4 micras, aumenta la resolución de las imágenes.
La tinción es la mayor diferencia con la histopatología convencional. Para la tinción se utilizan fluorocromos, moléculas que tienen la propiedad de responder a la radiación ultravioleta de determinada longitud de onda, emitiendo radiación ultravioleta de otra longitud de onda, generalmente en el espectro visible. El fluorocromo más utilizado es el isotiocianato de fluoresceína (FITC) que emite fluorescencia verde 41.
La clave de la inmunofluorescencia es la utilización de anticuerpos de especies no humanas conjugados con un fluorocromo y específicos contra inmunoglobulinas o complemento humano. Describimos a continuación las técnicas de marcaje directo e indirecto.
En la técnica directa, la más utilizada de forma rutinaria, se utiliza un anticuerpo primario antiinmunoglobulina humana (anti-IgG, anti-IgA, anti-IgM) o anticomplemento humano (anti-C3) de origen animal y conjugado generalmente con FITC.
El marcaje indirecto utiliza dos anticuerpos: primario y secundario. En el estudio de enfermedades ampollosas el anticuerpo primario es una IgG antiinmunoglobulina humana o anticomplemento humano generado en una especie no humana (ratón, cabra, conejo y otros). El anticuerpo secundario en una anti-IgG murina, anti-IgG caprina o lo que corresponda según el animal de origen, que sólo se une donde se haya unido previamente el anticuerpo primario. El anticuerpo secundario es el que está conjugado con el fluorocromo y su localización puede verse mediante iluminación con la longitud de onda específica l que estimula este fluorocromo (fig. 7). La técnica de marcaje indirecto aumenta la sensibilidad, pero reduce la especificidad al incrementar la tinción inespecífica.
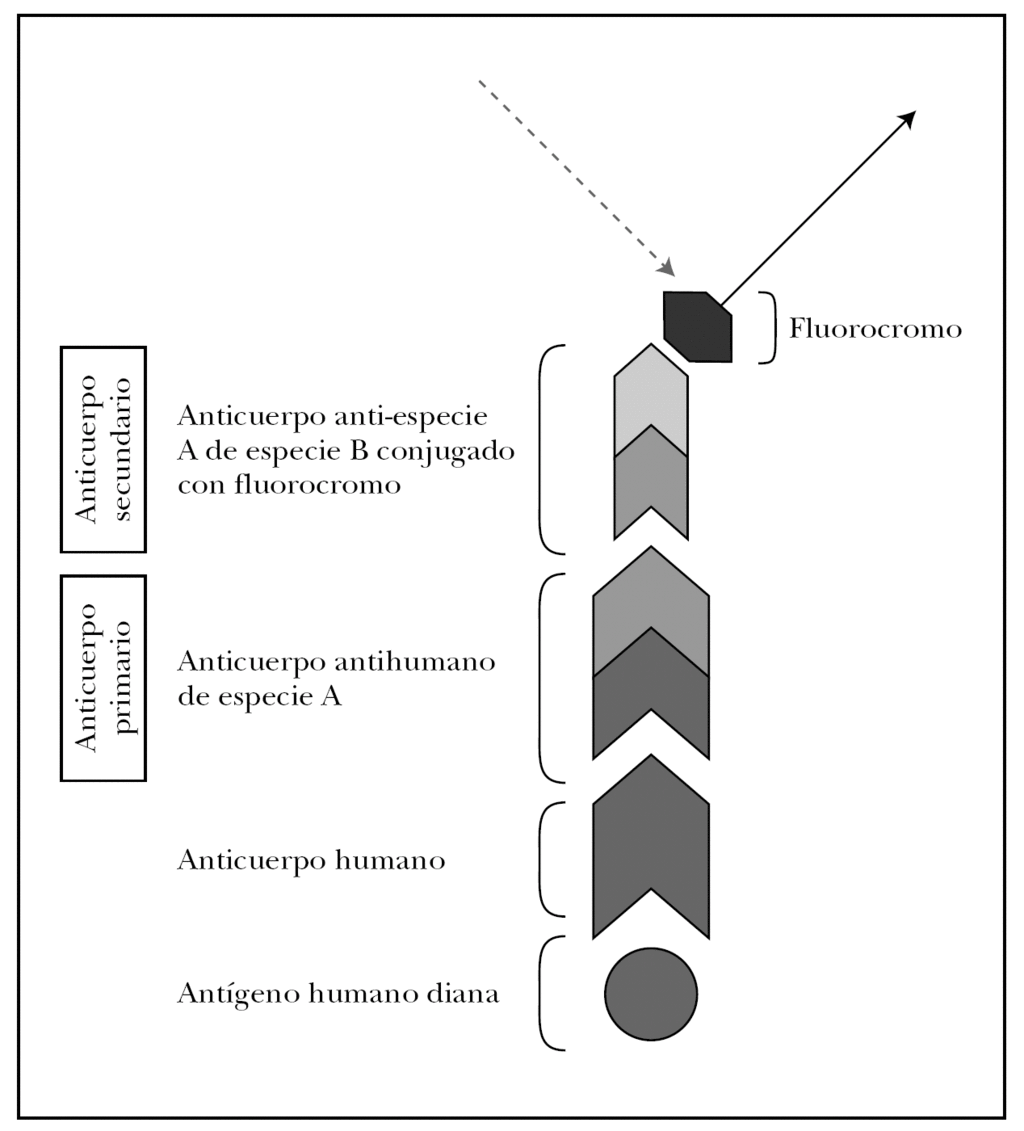
Fig. 7.--Fundamento de la inmunofluorescencia. Técnica de marcaje indirecto para inmunofluorescencia directa.
4. Diagnóstico. El microscopio de fluorescencia convencional tiene un manejo muy semejante al de los microscopios ópticos normales. Es importante ser rápido, ya que la fluorescencia va desapareciendo con el tiempo y la muestra «se quema». Cuando no se esté observando por el ocular debe interrumpirse la llegada de luz al cristal para alargar su vida.
En las enfermedades ampollosas subepidérmicas siempre hay depósitos localizados en la unión dermoepidérmica y en la dermatitis herpetiforme, además existen depósitos en las papilas dérmicas. Tras comprobar la positividad de la fluorescencia en la unión dermoepidérmica hay que analizar 4 puntos 38-41: a) tipo de depósito autoinmune: IgG, IgA, IgM, complemento; b) existencia de uno o varios tipos de depósitos; c) patrón de fluorescencia: fundamentalmente lineal o granular, y c) posible existencia de fluorescencia fuera de la unión dermoepidérmica.
Los principales patrones de depósito son el lineal y el granular. Existe un depósito lineal en el penfigoide ampolloso, penfigoide gestacional, penfigoide de mucosas, penfigoide anti-p200, dermatosis IgA lineal, epidermolisis ampollosa adquirida y lupus eritematoso ampolloso, o lo que es lo mismo, en todas las EASA excepto en la dermatitis herpetiforme, en que existen depósitos granulares.
Los epitelios foliculares y de glándulas sudoríparas pueden mostrar positividad. Hay que tener cuidado con los artefactos por papilas dérmicas que se pueden confundir con vasos.
Depósitos únicos en la membrana basal.
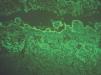
IgG y/o C3. Se ven en el penfigoide ampolloso, penfigoide de mucosas, penfigoide gestacional, liquen plano penfigoide, penfigoide anti-p200, epidermolisis ampollosa adquirida y lupus eritematoso ampolloso (fig. 8).
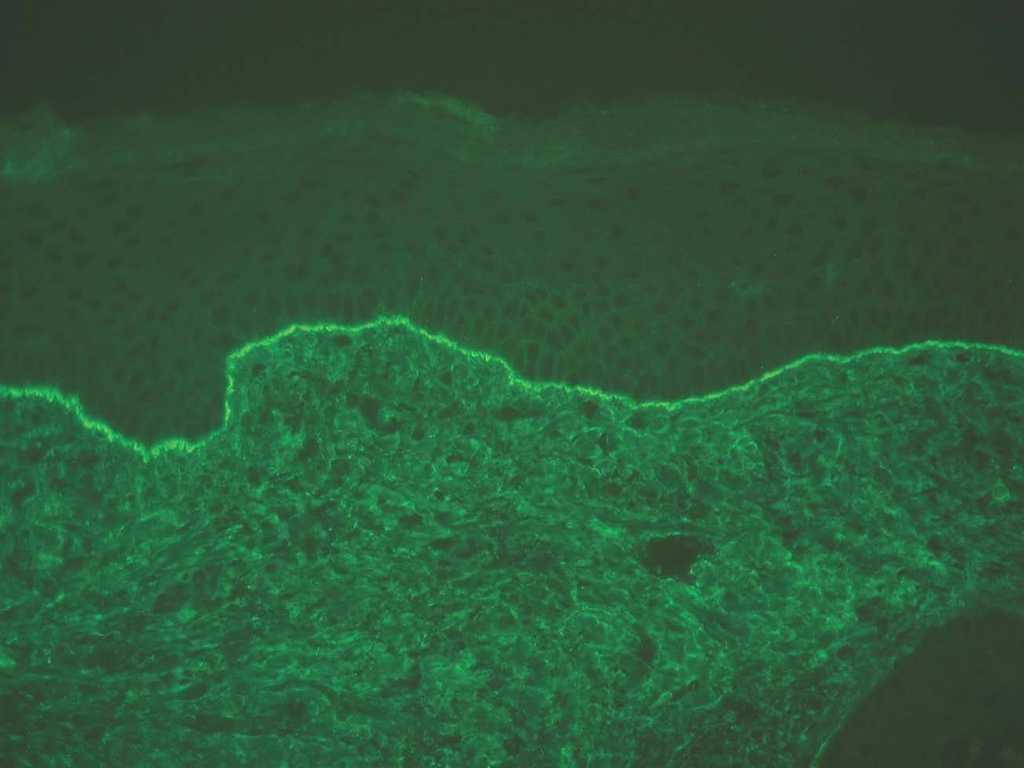
Fig. 8.--Depósito lineal de IgG en penfigoide ampolloso.
Si aparece un depósito de C3 único o más intenso que IgG es sugerente de la familia del penfigoide. Si aparece IgG más intenso que C3 son más probables epidermólisis ampollosas adquiridas (EAA) y lupus eritematoso ampolloso (LEA).
Vodegel et al 52 trataron de buscar mayor capacidad diagnóstica a la IFD y subdividieron el patrón lineal en patrón lineal puro y patrón en sierra. El patrón en sierra se divide asimismo en dos: en N y en U. Observaron que el patrón en U es exclusivo de las enfermedades del colágeno VII: EAA y LEA, mientras que el patrón en N ocurre en todas las demás (penfigoide ampolloso, penfigoide gestacional, penfigoide de mucosas, penfigoide anti-p200, y dermatosis IgA lineal). Para observar estos patrones en U y en N hay que realizar criosecciones muy finas, menores de 4 micras, que no suelen hacerse en la práctica rutinaria.
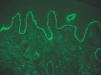
IgA: el depósito lineal de IgA caracteriza la dermatosis IgA lineal (fig. 9). El depósito de C3 es más infrecuente y menos intenso. La morfología de los depósitos es similar a los de penfigoide ampolloso y EAA. Como diagnóstico diferencial, algún penfigoide de mucosas puede depositar exclusivamente IgA y C3.
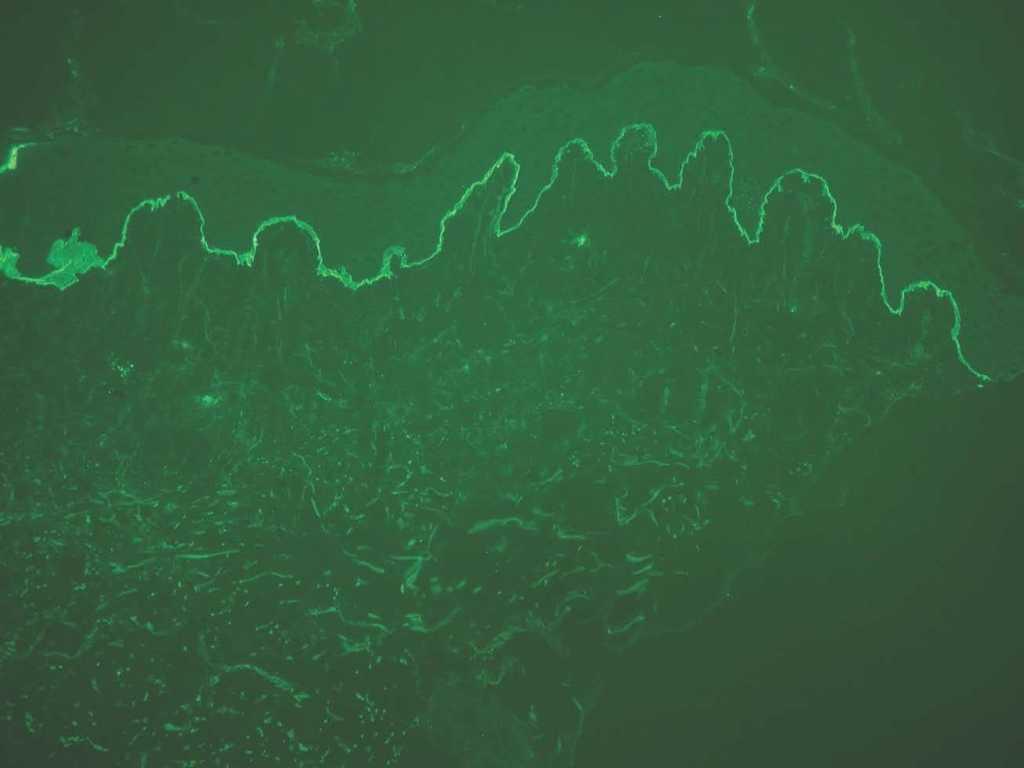
Fig. 9.--Depósito lineal de IgA en dermatosis IgA lineal.
C3: como ya se ha mencionado, un depósito único de C3 puede verse en penfigoide ampolloso, penfigoide gestacional, penfigoide de mucosas y liquen plano penfigoide (fig. 10).
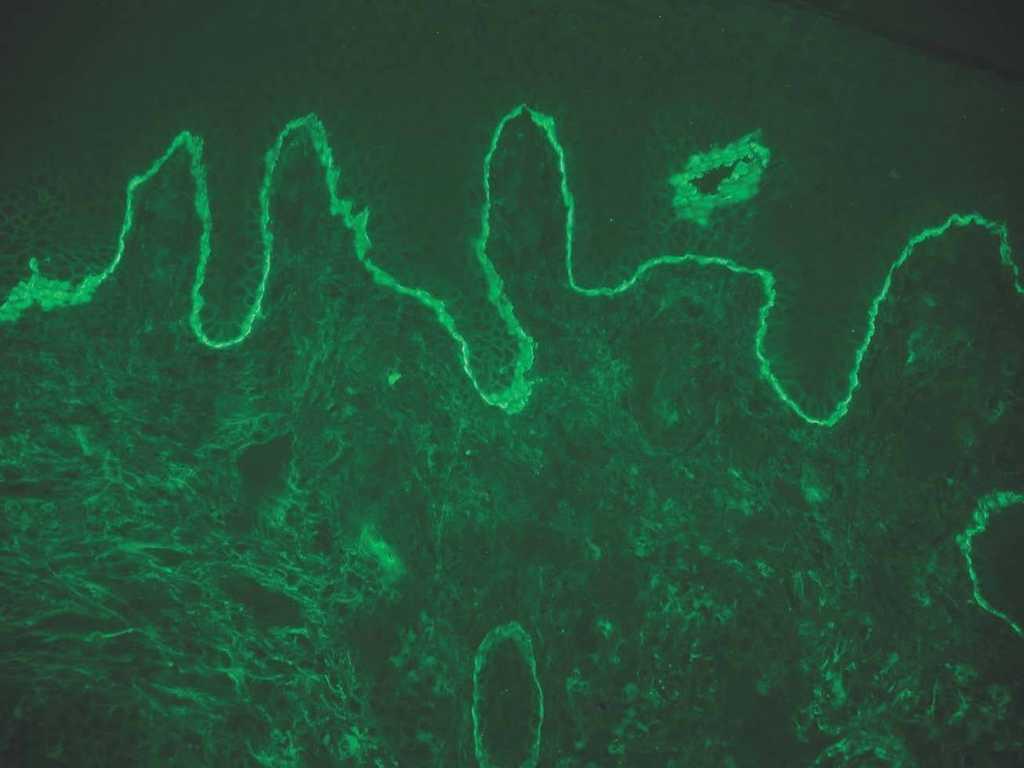
Fig. 10.--Intenso depósito de C3 en penfigoide ampolloso.
Depósitos múltiples en la membrana basal. Cuando se deposita más de una clase de inmunoglobulina son más probables EAA y LEA. En la EAA casi siempre hay IgG intensa, en la mitad de los casos hay IgM y en un tercio IgA.
En el LEA el 60 % tienen depósitos indistinguibles de la EAA. El resto tienen depósitos granulares continuos semejantes a los de algunos lupus no ampollosos. El depósito de IgA se asocia a LEA más que a lupus eritematoso no ampolloso.
Es posible la existencia de penfigoide de mucosas con IgG e IgA y dermatosis IgA lineal con IgA e IgG.
Depósito en papilas dérmicas. Como ya se ha mencionado, en la dermatitis herpetiforme se producen depósitos granulares de IgA y C3 en las papilas dérmicas y unión dermoepidérmica. La imagen es patognomónica. Se deposita IgA en el 100 % y C3 en el 50 %. Son posibles los depósitos IgG e IgM, pero con menos frecuencia e intensidad (fig. 11).
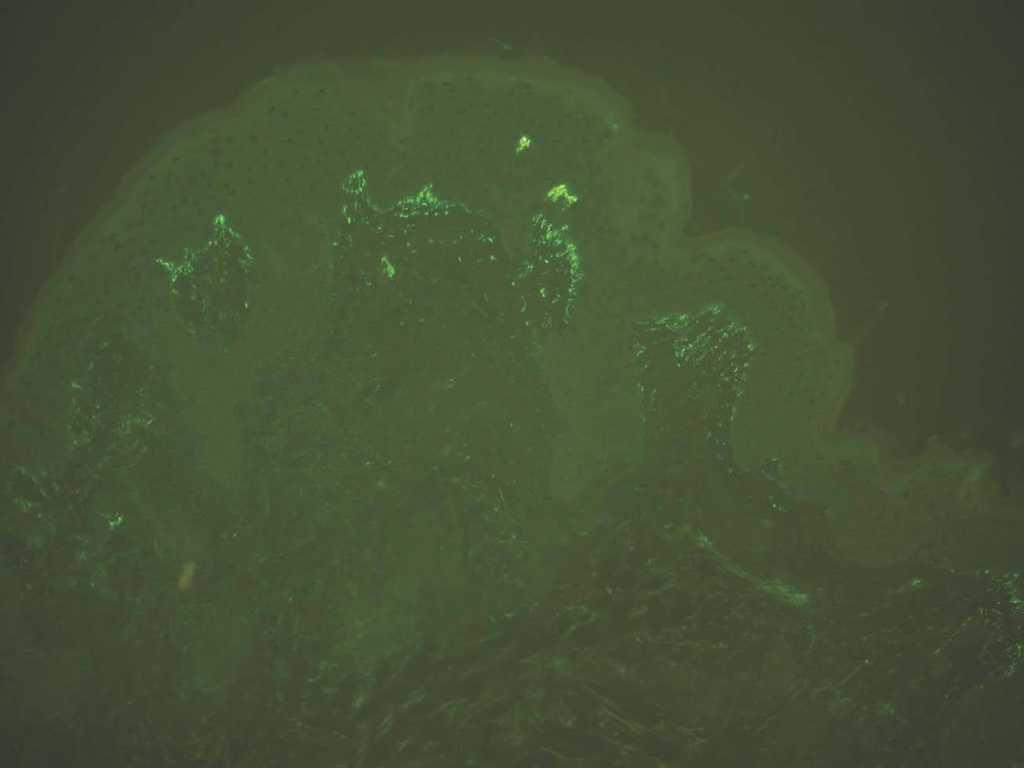
Fig. 11.--Depósito granular de IgA en dermatitis herpetiforme.
5. Almacenamiento. Se ha investigado la estabilidad de las preparaciones de inmunofluorescencia almacenadas a temperatura ambiente. En un estudio se comprobó que a los 12 meses eran visibles el 92 % de las preparaciones, pero a los 20 meses tan sólo el 28 %. Por tanto, es muy recomendable fotografiar los resultados y almacenarlos digitalmente 53.
Inmunofluorescencia directa sobre piel separada en NaCl 1M. La separación de la piel a lo largo de la unión dermoepidérmica a un nivel conocido es un ingenioso abordaje para diferenciar enfermedades. Se han publicado 4 métodos de separación: NaCl 1M, tripsina, creación de una ampolla por succión y activación de proteasas en salino tamponado con fosfatos. En todos estos métodos el colágeno XVII se mantiene en el lado epidérmico, mientras que la laminina y los colágenos IV y VII se mantienen en el lado dérmico 54 (tabla 1).
El método que más se ha extendido es la separación mediante NaCl 1M. La biopsia se incuba de 48 a 72 horas en NaCl 1M, a 4 °C y posteriormente se realiza la tinción habitual de inmunofluorescencia directa 55-57.
En general en el penfigoide ampolloso, penfigoide gestacional y penfigoide de mucosas los depósitos se ven en el lado epidérmico, convencionalmente superior o «techo» de la hendidura artificial (fig. 12). Los depósitos se corresponden con la localización del dominio extracelular del colágeno XVII y de NC16A, que contiene los epítopos más frecuentes reconocidos por los anticuerpos patogénicos. Como excepciones, en el penfigoide anti p-200 y en el penfigoide de mucosas anti-epiligrina o anti-laminina 5 los depósitos están en el lado dérmico.
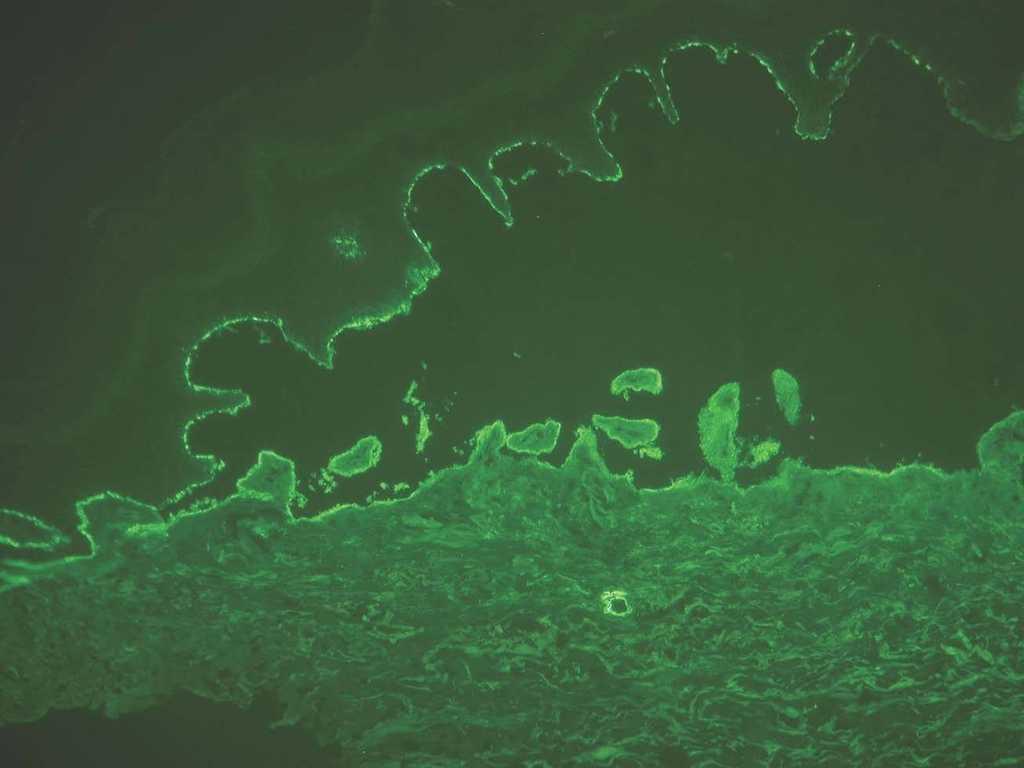
Fig. 12.--Inmunofluorescencia directa de piel separada en NaCl 1M: depósito en el lado epidérmico de separación en penfigoide ampolloso.
Los depósitos en la EAA y en el LEA se ven en el lado dérmico de la separación. Están situados en la sublámina densa, donde se sitúa el colágeno VII (fig. 13).
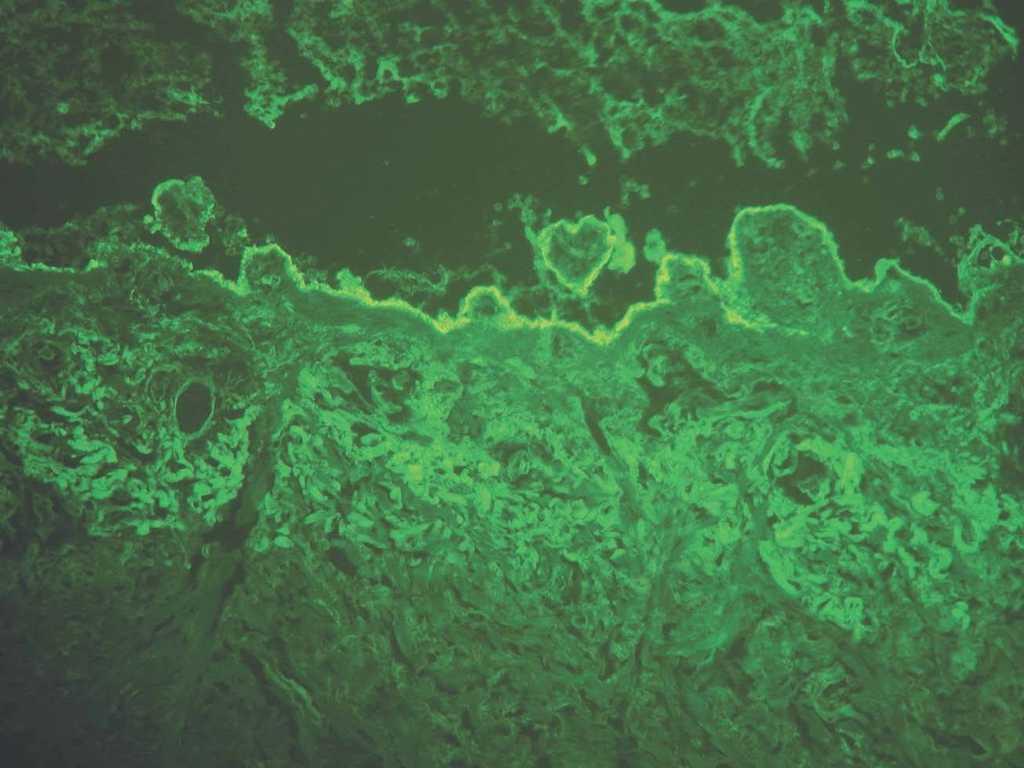
Fig. 13.--Inmunofluorescencia directa de piel separada en NaCl 1M: depósito en el lado dérmico de separación en epidermolisis ampollosa adquirida.
En la dermatosis IgA lineal es más frecuente la localización en el lado epidérmico, pero también hay una forma que marca en el lado dérmico, asociada a reactividad contra el colágeno VII.
Los hallazgos de inmunofluorescencia directa en cada enfermedad se resumen en la tabla 2.
Inmunofluorescencia directa mediante microscopio confocal. El término «confocal» designa un sistema óptico común a dispositivos muy distintos (microscopía convencional, microscopía de inmunofluorescencia y microscopía in vivo) que permite eliminar los artefactos por superposición al permitir el paso de la luz reflejada desde un único plano y eliminar la procedente de puntos fuera de foco. El efecto puede compararse al de un microtomo óptico 58-62.
El microscopio confocal espectral es un microscopio de fluorescencia sofisticado. En lugar de observar directamente a través de los oculares, un software procesa las imágenes y realiza reconstrucciones. En enfermedades ampollosas realiza cortes que permiten estudios de alta resolución de la morfología de los depósitos. El software permite analizar dos o más fluorocromos simultáneamente, realizar superposiciones de imágenes y visualizar el área de colocalización de proteínas seleccionadas. La colocalización de proteínas y anticuerpos puede visualizarse e incluso realizar un análisis numérico.
En el diagnóstico de enfermedades ampollosas mediante microscopía confocal se realiza una técnica denominada FOAM 63-71 (fluorescent overlay antigen mapping) cuyo objetivo es comparar la localización de los depósitos autoinmunes con antígenos conocidos de la membrana basal que elegimos como marcadores, tras realizar una doble tinción. Las proteínas marcadoras elegidas con mayor frecuencia son el colágeno VII, colágeno IV, laminina y subunidad β4 de integrina α6β4 (fig. 14).
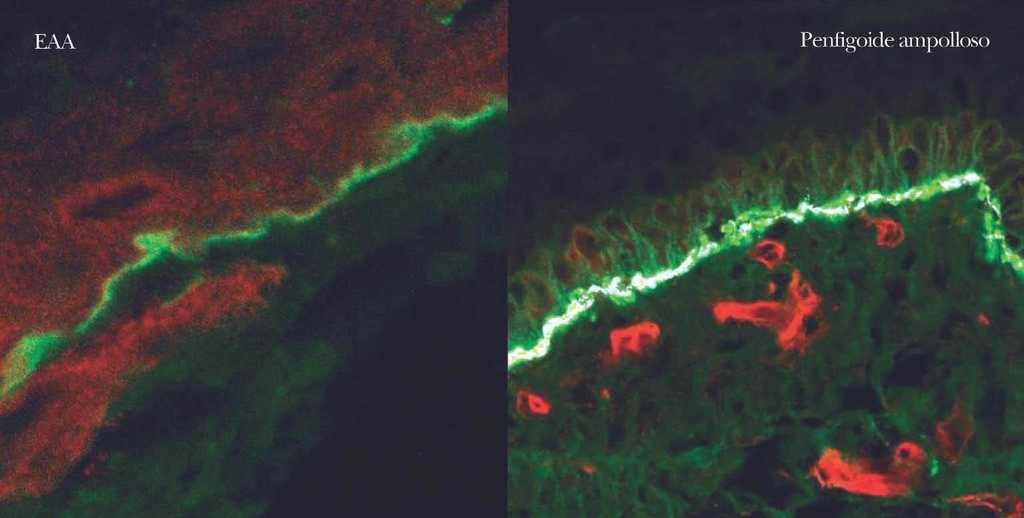
Fig. 14.--FOAM (fluorescent overlay antigen mapping) mediante microscopía confocal. Estudio de colocalización de β4 integrina con depósitos autoinmunes. En epidermolisis ampollosa adquirida (EAA) se observa una nítida banda verde de IgG situada inferior a la integrina (roja) en consonancia con la situación del colágeno VII. En un caso de penfigoide ampolloso se encuentra colocalización de β4 integrina con los depósitos autoinmunes, lo cual concuerda con autoinmunidad contra colágeno XVII.
Inmunomicroscopía electrónica
La inmunomicroscopía electrónica es una variante de la microscopía electrónica en la cual se marcan los depósitos autoinmunes con un material electrodenso. El concepto es similar al de la inmunofluorescencia directa con la diferencia de que el último marcador es oro, que resulta denso al haz de electrones 38,72.
En penfigoide ampolloso se ve en la mayoría de casos IgG en el lado extracelular de la membrana plasmática del queratinocito basal, bajo los hemidesmosomas, compatible con la localización del segmento NC16A del colágeno XVII.
Es una técnica muy poco utilizada para el diagnóstico.
Detección de autoanticuerpos en suero
Los autoanticuerpos contra las proteínas de la unión dermoepidérmica no se encuentran sólo en la piel. También es posible detectarlos en suero y orina 73. Se han desarrollado distintas técnicas para la detección y clasificación de estos autoanticuerpos que detallamos a continuación.
Inmunofluorescencia indirecta
Por inmunofluorescencia indirecta se entiende el conjunto de técnicas en las que el suero o exudado se incuba con un tejido sano con todas las estructuras moleculares conservadas 38,41,74,75. Se han usado como sustratos piel humana normal y esófago de mono. La sensibilidad y especificidad en cada caso depende del sustrato y del tipo de patología. Las imágenes obtenidas son semejantes a las de la inmunofluorescencia indirecta y se interpretan siguiendo los mismos criterios.
Inmunofluorescencia indirecta convencional. El suero del paciente se extrae por centrifugación. Se incuba el sustrato (tejido sano) con el suero problema 30 minutos. Tras lavado se incuba con antiinmunoglobulina humana marcada con un fluorocromo.
En una variante, poco usada, se incuba el sustrato-suero con complemento, y después con anti-C3 humano.
1. Depósitos de IgG. Se encuentran anticuerpos IgG anti-membrana basal en penfigoide ampolloso, penfigoide de mucosas, penfigoide gestacional, EAA y LEA, con prevalencia variable. En penfigoide gestacional sólo es positivo en el 10 %, en el penfigoide de mucosas en el 10-25 %. La sensibilidad sube al 60-80 % en penfigoides ampollosos y al 50 % en EAA. En el LEA sólo se detectan anticuerpos sobre piel separada (ver más adelante).
El patrón de fluorescencia no ayuda al diagnóstico diferencial. Los títulos de anticuerpos determinados mediante esta técnica no se correlacionan con la actividad en penfigoide ampolloso.
2. Depósitos de IgA. Se encuentran IgA anti-membrana basal en la dermatosis ampollosa IgA lineal entre un tercio y la mitad de los pacientes. La sensibilidad aumenta en piel humana separada en NaCl 1M (ver más adelante). Los títulos son bajos.
En dermatitis herpetiforme hay anticuerpos antiendomisio que se unen al músculo liso hasta en el 70 % de los pacientes que no siguen la dieta sin gluten.
En algunos casos de penfigoide ampolloso pueden detectarse IgA, siempre con IgG.
Inmunofluorescencia indirecta sobre piel separada. Una variante de inmunofluorescencia indirecta consiste en separar la piel sana en NaCl 1M, como en la IFD 41,76-80. Aumenta la sensibilidad en todas las enfermedades y es la única manera de visualizar anticuerpos en el LEA. En el penfigoide ampolloso la sensibilidad llega al 90 %.
En penfigoide ampolloso, penfigoide gestacional y muchos penfigoides de mucosas se visualizan anticuerpos en el techo de la hendidura artificial. En epidermolisis ampollosa adquirida, penfigoide de mucosas antiepiligrina, penfigoide anti-p200 y LEA la fluorescencia se localiza en el suelo de la hendidura. En dermatosis IgA lineal puede aparecer fluorescencia en cualquiera de los dos lados.
Inmunofluorescencia indirecta sobre sustratos deficitarios. Se trata de una técnica poco empleada en la que el suero se incuba con piel patológica deficiente en proteínas conocidas (colágeno XVII o colágeno VII). Si la autoinmunidad es, por ejemplo, contra colágeno VII y se trata de una piel deficiente en colágeno VII no se detectará unión. Esta técnica tiene grandes limitaciones 5,81.
Microscopía inmunoelectrónica indirecta. Consiste en estudiar bajo el microscopio electrónico piel sana incubada con suero problema, con los depósitos autoinmunes convenientemente marcados. En el penfigoide ampolloso se ve cómo se unen los anti-BP230 a la placa hemidesmosómica y los anti-colágeno XVII a lamina lúcida bajo hemidesmosomas en penfigoide ampolloso. En penfigoide de mucosas se ha visto densidad en lámina densa y lámina lúcida inferior.
En EAA se ven partículas de oro a lo largo de las fibrillas de anclaje.
ELISA
ELISA es un acrónimo de enzyme-linked immunosorbent assay (inmunoabsorción ligada a enzimas). Existen distintas variantes del ELISA según se quiera detectar antígenos o anticuerpos 82. Para detectar autoanticuerpos se emplea el ELISA indirecto, en el que los pocillos tienen el antígeno fijo. La técnica se observa en la figura 15 y se puede resumir en los siguientes pasos 82,83.
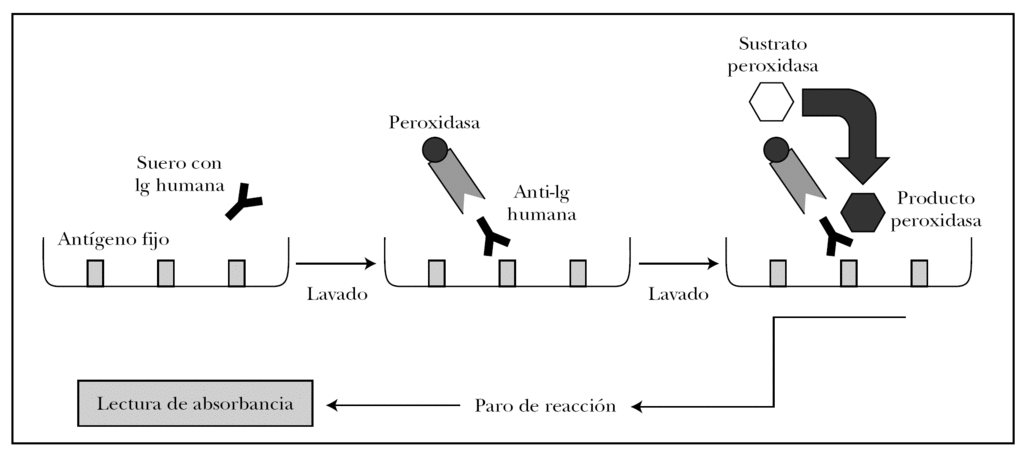
Fig. 15.--Técnica del ELISA indirecto.
1. Cada pocillo se incuba con diluciones de sueros problema y sueros control. Según la concentración de autoanticuerpos en el suero problema se producirá mayor o menor unión antígeno-anticuerpo, que se puede cuantificar.
2. Se añade como anticuerpo secundario antiinmunoglobulina humana de origen animal conjugada con peroxidasa.
3. El grado de unión anticuerpo secundario-anticuerpo primario se cuantifica tras lavado añadiendo un sustrato para la peroxidasa. Si ha habido mucha unión, habrá mayor cantidad de peroxidasa en el pocillo, y por tanto se producirá mayor producto de la reacción enzimática.
4. La reacción se para en un tiempo predeterminado.
5. La cantidad de producto es proporcional a la absorbancia del pocillo. Como ventajas, no utiliza antígenos desnaturalizados, es rápida, sensible y permite analizar múltiples sueros a la vez.
Existen varios kits comerciales. El más utilizado detecta anticuerpos contra el segmento NC16A del colágeno XVII. En la fase sólida de los pocillos hay NC16A de origen recombinante. En penfigoide ampolloso el ELISA contra NC16A tiene una sensibilidad del 94 % y especificidad del 97 %. En penfigoide gestacional la sensibilidad se acerca al 100 %. En cambio, el ELISA frente al colágeno XVII completo (también comercializado) sólo es positivo en el 66,6 % de los pacientes 84-89.
Se ha demostrado correlación de los títulos de anticuerpos contra NC16A con la actividad de la enfermedad y la respuesta terapéutica, lo que no ocurre con la inmunofluorescencia indirecta. Esto se atribuye a que la inmunofluorescencia indirecta detecta también anticuerpos contra BP230 junto a anticuerpos contra colágeno XVII 90-94.
Hay un ELISA para el dominio NC1 del colágeno VII, pero todavía no es comercial. Ha demostrado ser sensible y específico para EAA y LEA.
En dermatitis herpetiforme se realiza ELISA para detectar anticuerpos antitransglutaminasa y antiendomisio.
Inmunoblot
El término blot (mancha, borrón) se aplica a un conjunto de técnicas de biología molecular, en las que se realiza transferencia desde un gel de electroforesis a una membrana, donde se pueden visualizar los resultados o realizar incubaciones posteriores. El southern blot analiza ADN, el northern blot analiza ARN y el western blot analiza proteínas 95. Lo que se conoce como inmunoblot es un tipo de western blot en el que tras separar extractos proteicos de un tejido determinado, éstos se incuban con un suero problema que contiene potenciales autoanticuerpos 83,96,97.
El objetivo del inmunoblot en la patología ampollosa autoinmune es detectar contra qué proteínas dermoepidérmicas reaccionan anticuerpos en el suero.
El procedimiento tiene varias fases:
1. Purificación de extractos proteicos. Las proteínas pueden tener varios orígenes: extractos proteicos de piel sana (dérmicos o epidérmicos), extractos proteicos de cultivos celulares o proteínas recombinantes obtenidas en bacterias.
2. Electroforesis. Las proteínas se separan según su masa molecular mediante electroforesis en un gel de poliacrilamida.
3. Transferencia. Una vez separadas las proteínas, se transfieren del gel de poliacrilamida a una membrana de nitrocelulosa, también por un sistema electroforético.
4. Incubación. La membrana de nitrocelulosa se incuba con los sueros problema y los sueros control.
5. Revelado. Tras la incubación, los resultados pueden visualizarse por varios métodos. El de uso más extendido utiliza anticuerpos antiinmunoglobulina humana unidos a fosfatasa alcalina o peroxidasa y adición del sustrato enzimático adecuado. También se puede revelar mediante incubación con proteína A estafilocócica marcada con 125I y autorradiografía.
Los extractos dérmicos contienen el colágeno VII, sus fragmentos y el antígeno de 200 kDa identificado en el penfigoide anti-p200 97-105. Interesan para estudiar la epidermolisis ampollosa adquirida, LEA, algunos casos de dermatosis IgA lineal y el penfigoide anti-p200. Los extractos epidérmicos son útiles para el resto de enfermedades.
En penfigoide ampolloso el inmunoblot reconoce dos antígenos principalmente: uno de 230 kDA corresponde a BP230 y otro de 180 kDa que corresponde al colágeno tipo XVII. Dentro del colágeno XVII se reconoce con alta frecuencia el segmento NC16A y existen algunos casos de reconocimiento de LAD-1 (120 kDa). Hay casos esporádicos de reactividad a la plectina (500 kDa).
En penfigoide de mucosas se ha visto reactividad frente al colágeno XVII (NC16A, LAD-1 y BP180 4575), BP230, subunidades α6 y β4 de la integrina α6β4, laminina 5 (epiligrina) y laminina 6 106,107.
En la dermatosis IgA lineal se marca con más frecuencia LAD-1, aunque puede detectarse positividad contra otros segmentos del colágeno XVII y contra el colágeno VII.
En EAA sobre extractos dérmicos se detecta una banda positiva de 290 kDa y más raramente de 145 kDa, que corresponden a colágeno VII y su fragmento NC1 N-terminal. Hay dos casos con reactividad doble frente al colágeno VII y la proteína de 200 kDa identificada en el penfigoide anti-p200 que pueden considerarse EAA con fenómeno de expansión de epítopos.
La desventaja del inmunoblot es que al tener que desnaturalizar las proteínas, se pierden puntos de unión antígeno-anticuerpo denominados epítopos conformacionales. Esto hace bajar la sensibilidad de la técnica y aumenta el número de falsos negativos.
Inmunoprecipitación
Existe una técnica que no desnaturaliza las proteínas denominada inmunoprecipitación, que es más compleja y lenta que el inmunoblot y por tanto se usa fundamentalmente en investigación 83. Exige solubilizar cada proteína por separado y detectar, también por separado la precipitación de los posibles complejos antígeno-anticuerpo que se forman al poner en contacto extractos proteicos purificados y suero problema. Se ha utilizado más en el grupo del pénfigo. En enfermedades ampollosas subepidérmicas no resulta idónea para estudiar el colágeno XVII por tratarse de una proteína transmembrana difícil de solubilizar, pero ha demostrado utilidad en el penfigoide antiepiligrina para la detección de autoanticuerpos contra la laminina 5.
En la tabla 3 se resumen las ventajas y desventajas de las técnicas de inmunoblot, inmunoprecipitación y ELISA.

PROTOCOLO DE DIAGNOSTICO Y SEGUIMIENTO
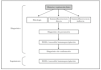
En la figura 16 mostramos el protocolo ideal de diagnóstico de las EASA 3. Es obvio que en la práctica clínica habitual de nuestro medio no son realizables muchas de las pruebas, que deben enviarse a centros de referencia nacionales e incluso internacionales. Es muy importante la colaboración interdisciplinar con los Servicios de Inmunología y Anatomía Patológica, e incluso la creación de Unidades de Enfermedades Ampollosas. Previsiblemente la técnica que presentará un mayor crecimiento en los próximos años será el ELISA, con la aparición de nuevos kits comerciales contra más antígenos de los ahora disponibles.
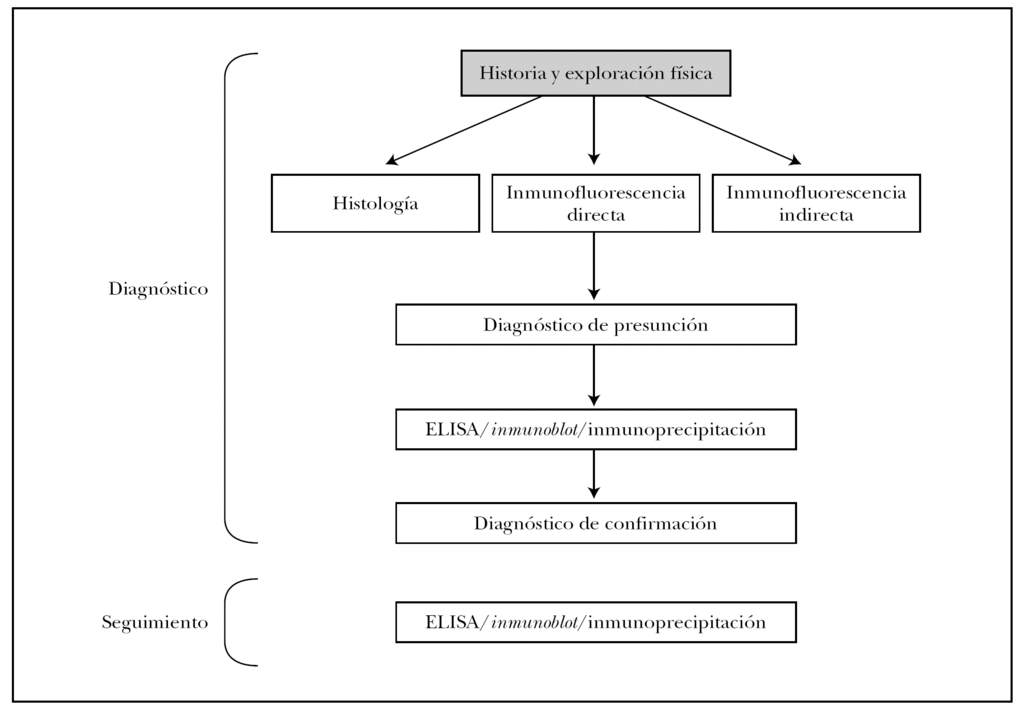
Fig. 16.--Protocolo diagnóstico de las enfermedades ampollosas subepidérmicas autoinmunes.
IMPLICACIONES TERAPÉUTICAS DE LOS RESULTADOS DIAGNOSTICOS
La importancia del diagnóstico preciso de las EASA reside en el diferente pronóstico que tienen las distintas enfermedades y su diferente abordaje terapéutico. Por ejemplo, la EAA tiene una característica resistencia a los corticoides orales que la diferencia del penfigoide ampolloso, enfermedad con inmunofluorescencia directa casi idéntica.
Aunque todavía no muy generalizado, el control de la actividad de la enfermedad de las EASA mediante ELISA permitirá un mejor ajuste de la medicación inmunosupresora y el tratamiento precoz de los rebrotes.
AGRADECIMIENTOS
Agradecemos al Dr. Manuel Lecona las imágenes histopatológicas y de inmunofluorescencia. Las imágenes de microscopía confocal han sido realizadas en la Unidad de Microscopía Confocal del Hospital General Universitario Gregorio Marañón, coordinada por la Dra. Paloma Sánchez-Mateos. Las tinciones para microscopía confocal han sido realizadas por Isabel Treviño y la obtención de las imágenes ha sido posible gracias a Rafael Samaniego.
GLOSARIO
Confocal: adjetivo que se aplica a sistemas ópticos diversos que tienen en común el paso de la luz emitida por la muestra a través de un sólo punto, de forma que sólo se analiza la correspondiente a un plano determinado.
Dominio: parte de una proteína con estructura terciaria característica que en muchos casos determina una función o ligando.
Epítopo: conjunto de aminoácidos reconocido por un determinado anticuerpo determinado dentro de un dominio de una proteína.
Epítopo conformacional: conjunto de aminoácidos que son reconocidos por un determinado anticuerpo gracias a que la estructura terciaria de la proteína los sitúa en una organización tridimensional determinada.
Extremos C-terminal y N-terminal: toda proteína se forma por la unión de aminoácidos entre sí a través de sus grupos carboxilo y amino. Existen dos aminoácidos en los extremos de la cadena, uno con un grupo carboxilo libre (extremo C-terminal o carboxi-terminal) y otro con un grupo amino libre (extremo N-terminal o amino-terminal).
Fluorocromo: molécula que tiene la capacidad de emitir luz tras recibir el estímulo de luz de una longitud de onda determinada.
Glucoproteína: proteína por modificación enzimática que tiene unidos al núcleo polipeptídico residuos hidrocarbonados que intervienen en funciones de reconocimiento y adhesión.
Heterodímero: proteína formada por dos subunidades diferentes entre sí, normalmente provenientes de distintos genes.
Heterotrímero: proteína formada por 3 subunidades de las que al menos 2 son químicamente diferentes.
Homotrímero: molécula formada por tres subunidades de estructura química idéntica.
Inmunoblot: tipo de western blot encaminado a la detección de anticuerpos.
kDa (kilodalton): unidad de masa molecular equivalente a 1.000 dalton; 1 dalton es 1/12 de la masa de un átomo de carbono 12 y es aproximadamente la masa de un átomo de hidrógeno (1.660 538 86 (28) x 1027 kg).
Polipéptido: cadena de aminoácidos.
Membrana basal: tipo de matriz extracelular especializada que separa todos los epitelios del tejido conectivo subyacente.
Membrana plasmática basal: parte de la membrana plasmática de una célula de cualquier epitelio que se sitúa en el lado de la membrana basal, opuesto a la luz.
Secuencias tipo colágeno: las proteínas de la familia del colágeno se caracterizan por la repetición de los tripletes Gly-X-Y. La glicina es un aminoácido con un radical hidrófobo que confiere al colágeno la propiedad de ensamblarse formando triples hélices.
Subunidad: dentro de una proteína, cada una de las cadenas polipeptídicas que, asociadas entre sí, conforman su estructura cuaternaria.
Porción transmembrana: conjunto de aminoácidos de una proteína destinados a atravesar la membrana plasmática y conectar los fragmentos intracelulares y extracelulares.
Correspondencia:
Minia Campos Domínguez.
Dr. Esquerdo, 46. 28007 Madrid. España.
miniacampos@gmail.com
Recibido el 21 de agosto de 2006.
Aceptado el 4 de septiembre de 2006.


