














En esta serie de 2 artículos realizamos una revisión de las principales entidades dermatopatológicas que cursan con granulomas. Esta primera parte se ha centrado en la aclaración de los conceptos, la presentación de los tipos de granulomas y de las células gigantes, así como en entidades muy diversas de origen no infeccioso. Algunas de ellas de origen metabólico, como la necrobiosis lipoídica: otras relacionadas con linfomas, como la micosis fungoides granulomatosa, y otras tan extendidas que casi resultan un problema cotidiano en las consultas de dermatología, como la rosácea.
This series of 2 articles on dermatopathologic diagnoses reviews conditions in which granulomas form. Part 1 clarifies concepts, discusses the presentation of different types of granulomas and giant cells, and considers a large variety of noninfectious diseases. Some granulomatous diseases have a metabolic origin, as in necrobiosis lipoidica. Others, such as granulomatous mycosis fungoides, are related to lymphomas. Still others, such as rosacea, are so common that dermatologists see them nearly daily in clinical practice.
En dermatopatología, definimos como granuloma una colección más o menos compacta de macrófagos. Los macrófagos son células que proceden de los monocitos de la médula ósea. Estos últimos son células mononucleadas circulantes. Se dirigen a los tejidos, pasando a constituir células tisulares con capacidad macrofágica (macrófagos o histiocitos).
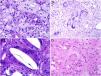
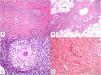
Los histiocitos de un granuloma suelen tener un citoplasma abundante, por lo que las células se parecen a las epiteliales. Este aspecto suele enfatizarse hablando de histiocitos o granulomas epitelioides. En muchas ocasiones, los histiocitos están tan próximos entre sí que no se pueden distinguir bien los límites celulares, pareciendo más bien un sincitio. Por ello, estrictamente hablando, las distribuciones dispersas de macrófagos, no se consideran granulomas. Si los histiocitos acumulan suficiente cantidad de material lipídico, pueden adoptar un citoplasma espumoso. Se suele hablar en ese caso de célula xantomatosa (fig. 1A).
A) Células xantomatosas. Se trata de histiocitos con un alto contenido citoplásmico lipídico, lo que les confiere un aspecto multivesicular (H&E ×400). B) Células de Langhans. Muestran múltiples núcleos polarizados a un extremo de la célula con una distribución en «herradura» (H&E ×400). C) Células gigantes de tipo cuerpo extraño. Dos de ellas aparecen abrazando un espacio acicular vacío correspondiente a un cristal de colesterol. Ambas muestran muchos núcleos en disposición desordenada (H&E ×400). D) Célula gigante de tipo Touton. Muestra los núcleos distribuidos en un círculo. El citoplasma interior a los círculos es densamente eosinofílico, mientras que la franja externa presenta un aspecto xantomatoso (H&E ×400).
En muchos granulomas, los mecanismos frustrados de fagocitosis pueden conllevar la aparición de histiocitos multinucleados de gran tamaño, conocidos como células gigantes multinucleadas. Algunas variantes de células gigantes tienen características histopatológicas peculiares, y aparecen con más frecuencia en ciertos tipos de granulomas, por lo que conviene conocerlas:
- -
La célula de Langhans muestra los núcleos desplazados a un polo de la célula en disposición como en herradura, con un citoplasma eosinofílico denso (fig. 1B).
- -
La célula de cuerpo extraño es una célula con muy abundante cantidad de núcleos (muchas veces más de 10), en disposición desordenada. No es raro que estas células aparezcan englobando parcialmente y adaptando su morfología a partículas del cuerpo extraño frente a las que se ha producido la reacción (fig. 1C).
- -
La célula de Touton es una célula con disposición circunferencial de los núcleos. Entre los núcleos y la membrana plasmática suele quedar un reborde periférico de citoplasma conservado que tiene un aspecto espumoso, a diferencia de las áreas centrales del citoplasma, que son densamente eosinofílicas (fig. 1D). Este tipo celular suele encontrarse en las respuestas inflamatorias con alto contenido lipídico.
Reconocemos distintos tipos de granulomas (fig. 2), que pueden tener distintos tipos de células gigantes. Ninguno de estos tipos de granulomas es patognomónico de ninguna entidad, aunque cada uno de ellos se asocia preferentemente a una o varias entidades.
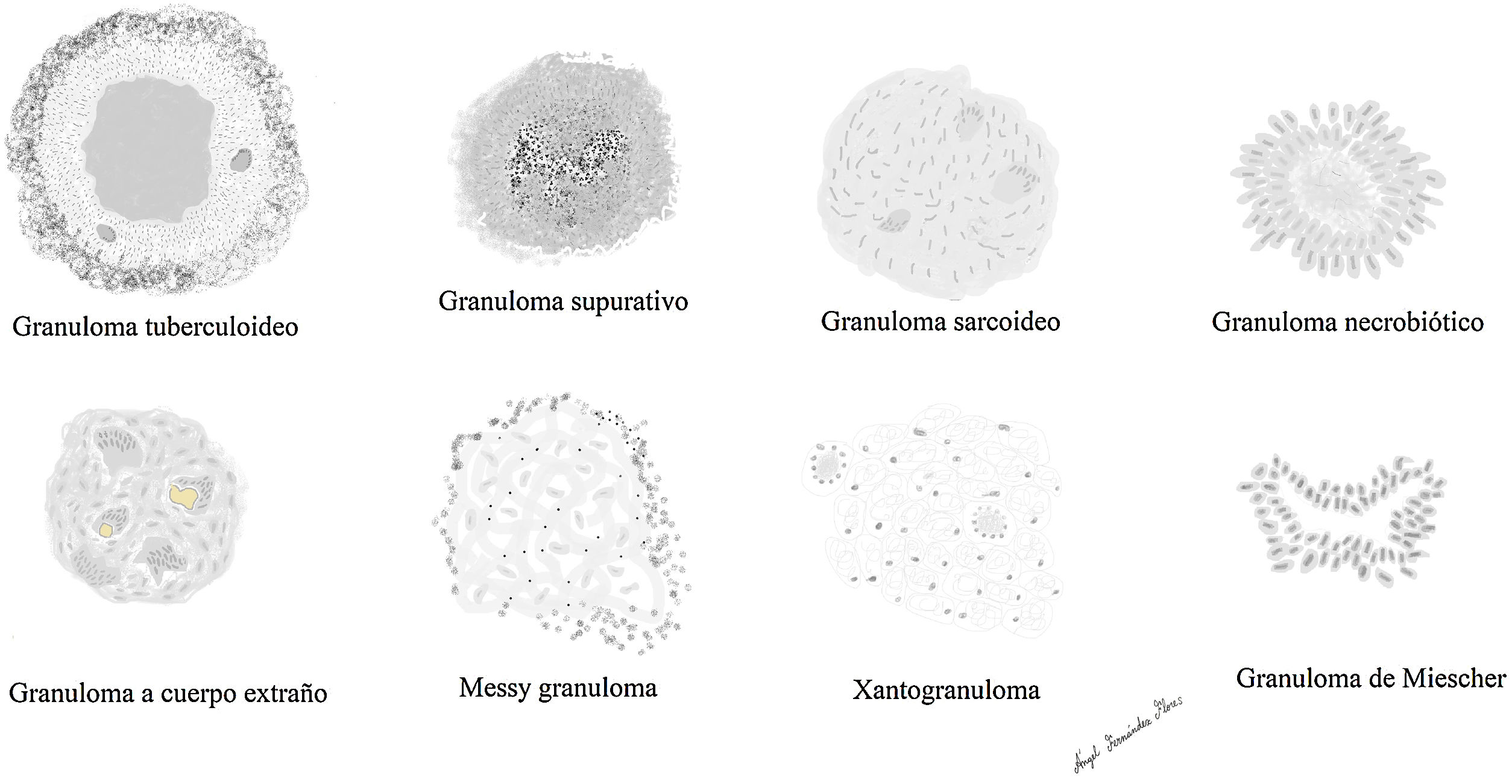
Los principales tipos de granulomas que reconocemos, son:
- -
Granuloma tuberculoideo
- -
Granuloma sarcoideo
- -
Granuloma supurativo o abscesificado
- -
Granuloma a cuerpo extraño
- -
Xantogranuloma
- -
Messy granuloma
- -
Granuloma necrobiótico
- -
Granuloma de Miescher
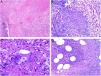
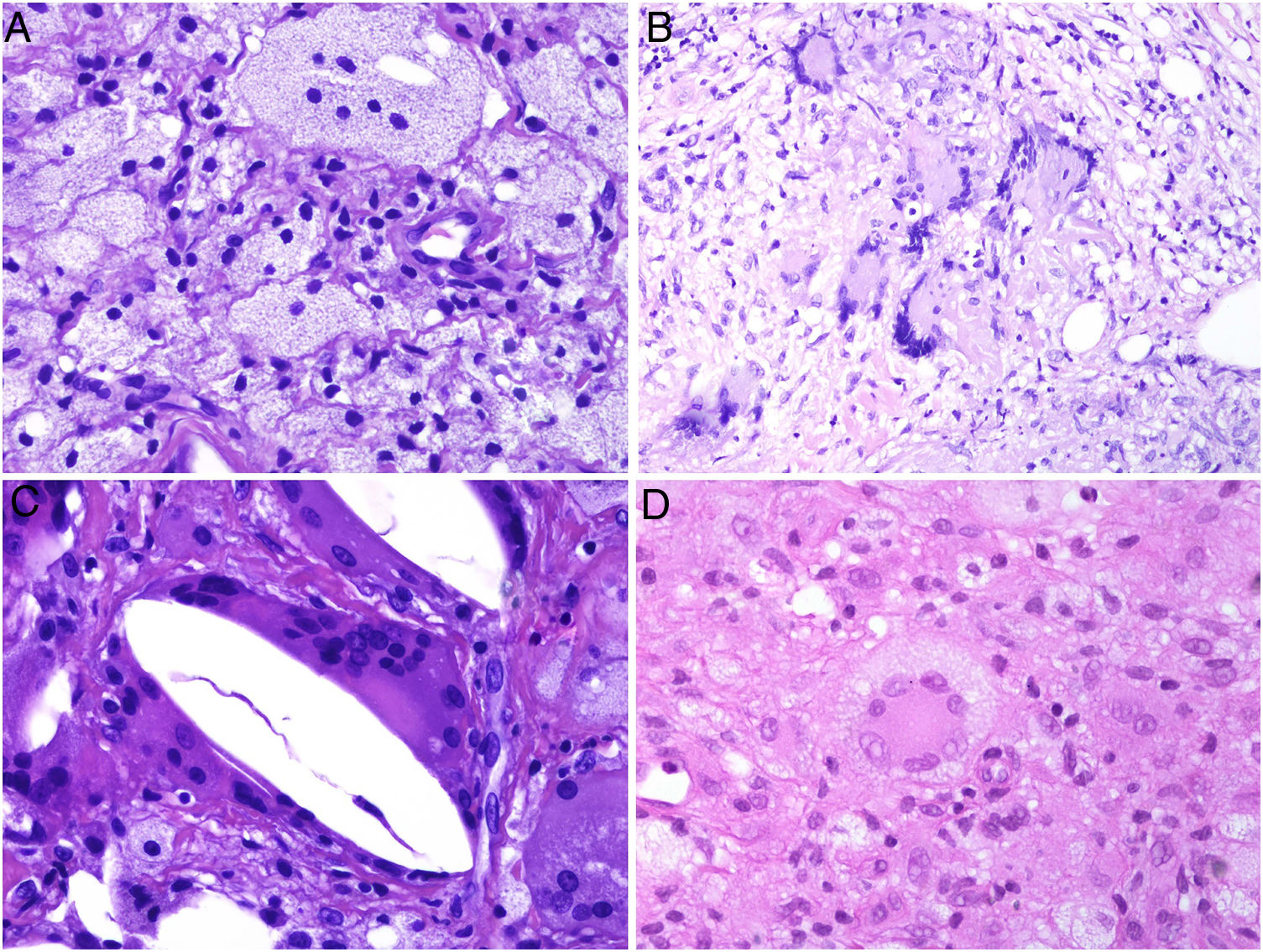
El granuloma tuberculoideo es un granuloma epiteliode que habitualmente muestra necrosis central de tipo caseoso. Esta última es al fin y al cabo necrosis coagulativa de los tejidos y, por lo tanto, amorfa y casi homogénea. El nombre caseoso le viene históricamente de su comparación con el caseum del requesón en el pasado. Suele presentar un infiltrado linfocitario prominente en la periferia del granuloma, que a veces se compara con una «corona» (fig. 3A). En muchas ocasiones pueden verse células gigantes del tipo Langhans. Este es el granuloma más frecuentemente asociado a la tuberculosis.
A) Granuloma tuberculoide. Muestra centro de necrosis caseosa rodeado por los histiocitos epitelioides y una evidente célula gigante de tipo Langhans. En periferia se observa una corona linfocitaria (H&E ×100). B) Granuloma sarcoideo. Granuloma epitelioide no necrosante y sin corona linfocitaria prominente («desnudo») (H&E ×100). C) Cuerpo asteroide. Muestra forma de estrella y ubicación intracitoplásmica. Es típico de los granulomas sarcoideos aunque no patognomónico (H&E ×400). D) Granuloma abscesificado (supurado) de una micobacteriosis atípica. Muestra histiocitos epitelioides rodeando un centro de polimorfonucleares neutrófilos (H&E ×100).
El granuloma sarcoideo, en contraposición, es un granuloma sin una prominente necrosis central. Cuando hay necrosis, es discreta y de tipo fibrinoide. Esta última se debe a depósito de fibrina junto con otros componentes como por ejemplo anticuerpos. Además, el infiltrado inflamatorio en la periferia del granuloma, no suele ser prominente. Por eso se dice que son granulomas «desnudos» (fig. 3B). Este es el granuloma que se suele presentar, por ejemplo, en la sarcoidosis. En esta última entidad, a veces se encuentran células gigantes con 2 tipos de inclusiones intracitoplásmicas peculiares. Los cuerpos asteroides tienen morfología de estrella (fig. 3C). Los cuerpos de Schaumann con cuerpos proteicos calcificados redondeados, en laminaciones concéntricas. Ninguna de estas 2 inclusiones es patognomónica, y también pueden verse en otros tipos de granulomas.
El granuloma supurativo o abscesificado muestra abundantes polimorfonucleares neutrófilos en su centro (fig. 3D). Este granuloma se suele ver con más frecuencia en las infecciones por hongos o por micobacterias atípicas.
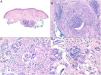
El granuloma a cuerpo extraño es morfológicamente muy variado. Eso es debido a que los cuerpos extraños frente a los que se puede desencadenar esta respuesta. Muy frecuentemente, las células gigantes aparecen abrazando y amoldándose a las partículas de cuerpo extraño que intentan fagocitar (fig. 4A). El granuloma se compone de histiocitos y respuesta linfocitaria, muchas veces sin una forma constante o predefinida.
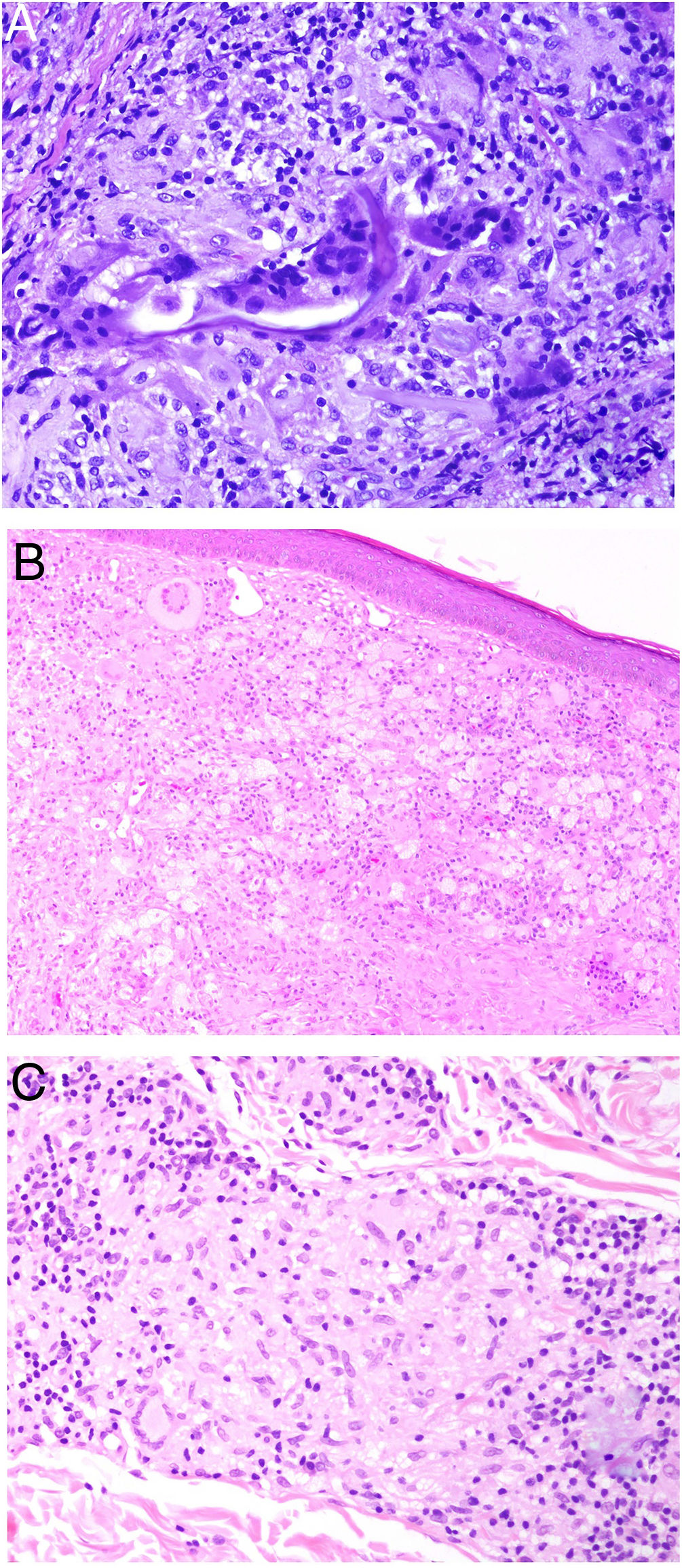
A) Granuloma a cuerpo extraño. Muestra numerosas células gigantes multinucleadas, de núcleos desordenados, abrazando el cuerpo extraño (H&E ×200). B) Xantogranuloma. Abundantes células espumosas junto con alguna ocasional célula de Touton (H&E ×40). C) Granuloma messy de una leishmaniosis. Muestra disposición desordenada de histiocitos de citoplasma claro y muy separados entre sí (H&E ×100).
El xantogranuloma es un granuloma principalmente constituido por histiocitos y células gigantes con cambios xantomatosos, es decir, con cambios citoplásmicos espumosos por acúmulo de componentes lipídicos (fig. 4B).
El messy granuloma (del inglés messy: desordenado) es un granuloma constituido por histiocitos epitelioides distribuidos laxamente y de modo desordenado (en distintas direcciones)1 (fig. 4C). El aspecto laxo es debido al citoplasma amplio y vidrioso de los histiocitos. Estos aparecen, además, separados por una sustancia hialina que presenta un color verdoso con tricrómico de Masson. El granuloma aparece permeado por linfocitos, y suele tener una corona linfocitaria, por lo que no se trata de granulomas «desnudos». Este tipo de granuloma es característico de la leishmaniasis.
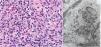
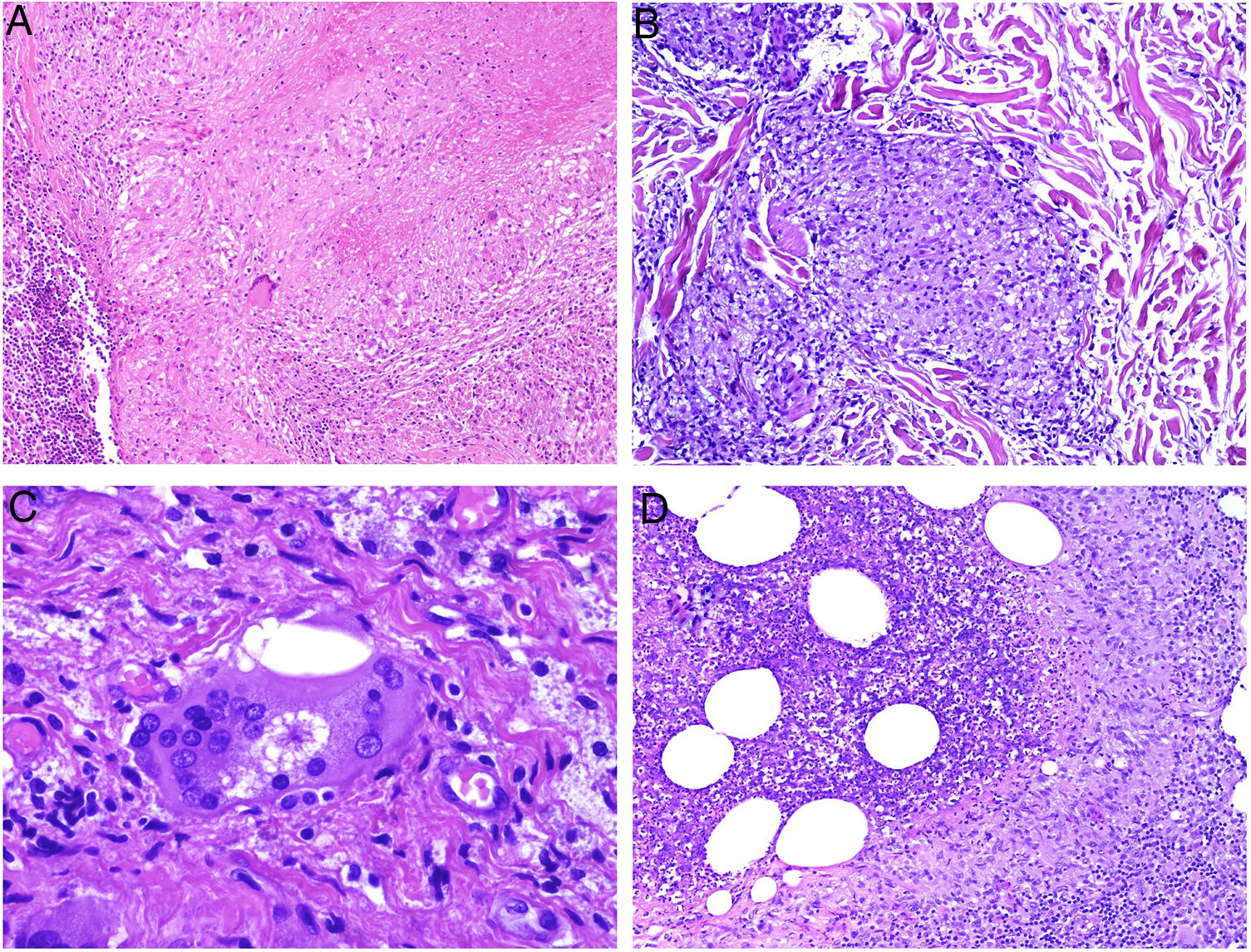
El granuloma necrobiótico define etimológicamente a un granuloma con tejido «muerto» en su centro. Esta definición es controvertida por varios motivos. En primer lugar, porque el material del centro de este grupo de granulomas está representado por tejido conectivo degenerado: fibras elásticas o colágenas, mucina… Al ser fibras conectivas, no son células y, por lo tanto, no pueden «morir» ni considerarse tejido muerto o vivo. Puede ocurrir (y de hecho es lo que sucede) que sus propiedades degeneren o se alteren. En segundo lugar, porque algunos de los granulomas no incluidos en esta categoría sí que podrían representar verdaderos granulomas necrobióticos (por ejemplo, el granuloma con necrosis central caseosa, en el que el material necrótico proviene de la muerte de células). Se incluyen en este grupo el granuloma anular, el xantogranuloma necrobiótico, los nódulos reumatoides o la necrobiosis lipoídica, entre otros. Dada la controversia con la denominación de necrobiótico, nos parece más adecuado denominar a este granuloma como degenerativo extracelular. No es raro que los histiocitos que rodean al centro degenerativo conectivo aparazecan dispuestos en empalizada, por lo que este granuloma también se ha conocido en la literatura como granuloma «en empalizada» (fig. 5A).
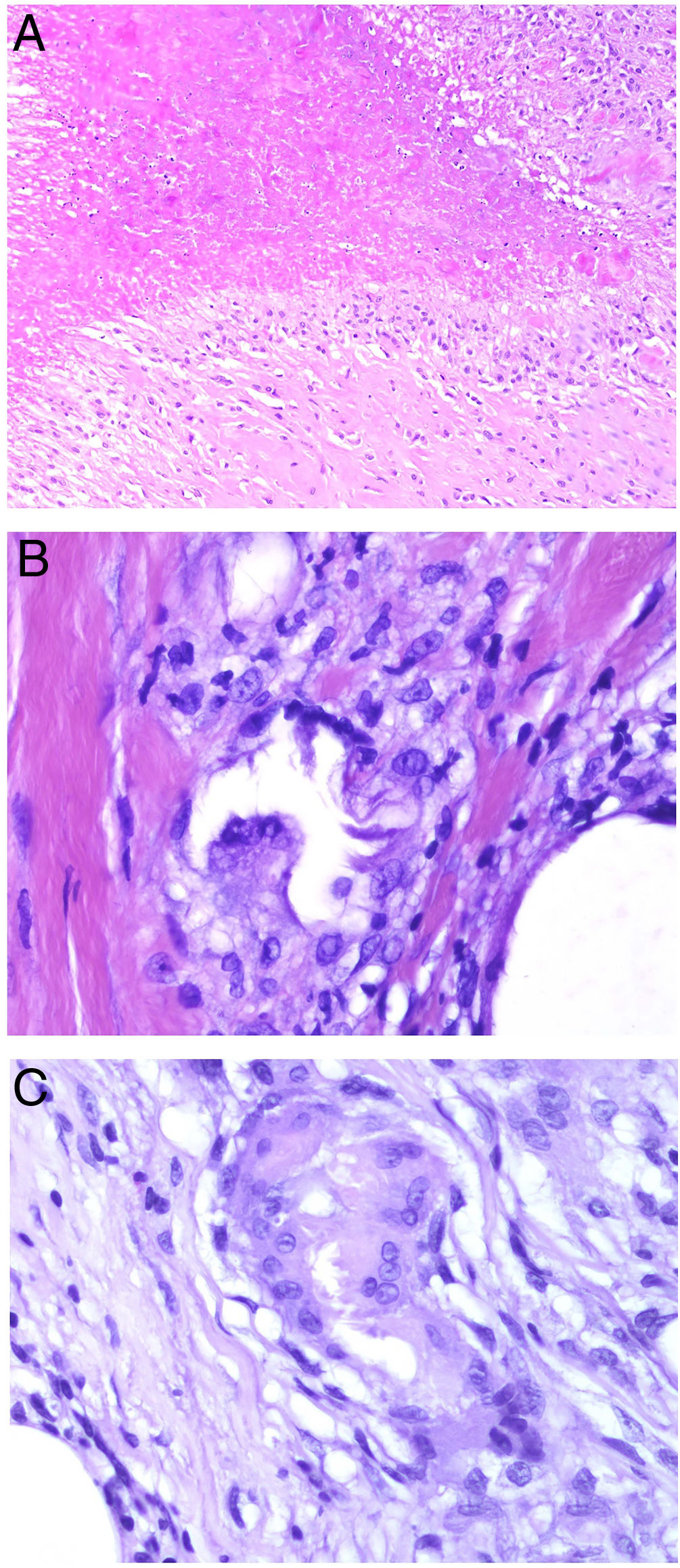
A) Granuloma necrobiótico (en empalizada) correspondiente a un nódulo reumatoide. Muestra material necrobiótico central con los histiocitos dispuestos en empalizada (H&E ×40). B) Granuloma de Miescher correspondiente a un eritema nodoso. Muestra espacio central angulado con histiocitos en periferia (H&E ×400). C) Granuloma de Miescher evolucionado. Con el tiempo, los granulomas de Miescher pueden estar constituidos casi exclusivamente por histiocitos multinucleados (H&E ×400).
El granuloma (radial) de Miescher muestra una colección de histiocitos pequeños de citoplasma pálido, dispuestos en torno a una hendidura central (fig. 5B). Con la evolución de estos granulomas, pueden aparecer algunas células gigantes en su periferia. La hendidura central aparece bien delimitada por una cutícula. Las células gigantes pueden llegar a ser el componente predominante o único del granuloma (fig. 5C), preservándose en su centro las estructuras hendidas con cutícula. Se desconoce la naturaleza de dichas hendiduras. Este granuloma es bastante específico del eritema nodoso2.
Entidades dermatopatológicas con respuesta granulomatosaA continuación, describiremos en 2 artículos, las principales entidades que conocemos en dermatopatología y que se caracterizan por incluir una respuesta granulomatosa prominente.
Reacciones a materiales de implante cosméticoDefinición: La inyección dérmica o subcutánea de materiales de relleno para corregir defectos cosméticos puede generar reacciones granulomatosas adversas histológicamente características del material implantado3.
Clínica: La apariencia clínica va desde tumefacción o eritema a ulceración o fistulización. Puede aparecer de forma temprana o tardía. Esta última es usualmente causada por productos no biodegradables, y puede asociarse a esclerosis o desplazamiento3,4.
Histopatología: Los patrones son muy variados e incluyen granulomas de tipo cuerpo extraño, empalizadas de histiocitos rodeando colecciones de material inyectado y reacciones granulomatosas intersticiales, sarcoideas o supurativas. A veces se acompañan de eosinófilos. En muchas ocasiones es posible reconocer el material inyectado o su silueta (por ejemplo, cavidades en «queso suizo», esferas o estructuras poligonales translúcidas, esferas azules, espículas polarizables o agregados de material azulado)5. Una de las más frecuentes es la reacción a silicona, caracterizada por la presencia de histiocitos con vacuolas de tamaño variable conteniendo material translúcido (seudolipoblastos) y frecuentes cuerpos asteroides. El ácido hialurónico, el más usado en la actualidad, aparece como lagunas de aspecto mucinoso (azul alcián positivo) entremezcladas con histiocitos, y puede conducir a fibrosis o encapsulación6 (fig. 6). No es infrecuente la detección de 2 materiales, ya sea porque forman parte de un mismo compuesto o porque un segundo implante activa un material que permaneció inerte durante años4.
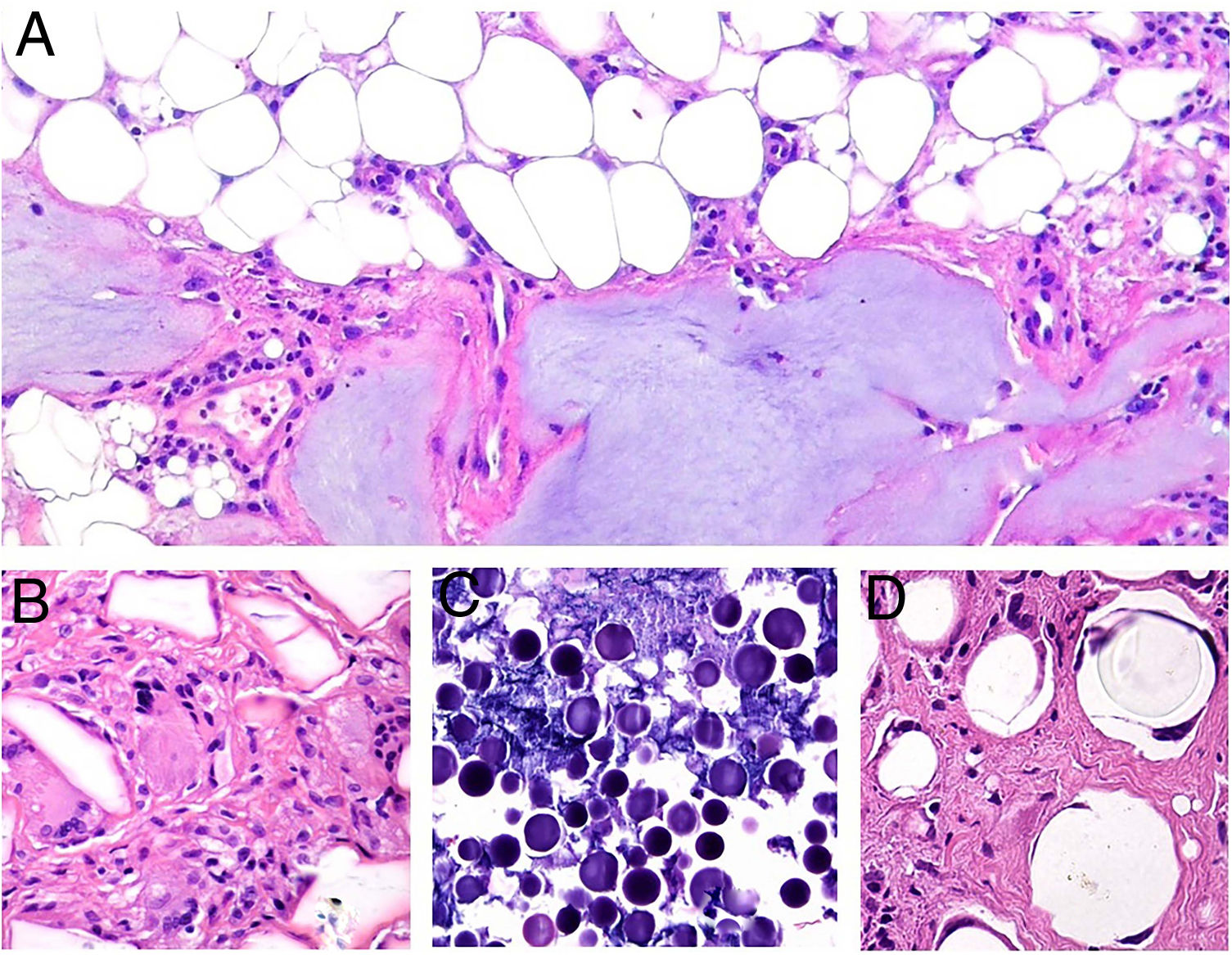
Granulomas a materiales utilizados en dermoestética. A) El material azulado de aspecto mixoide o mucinoso es ácido hialurónico. Está entremezclado con histiocitos conteniendo vacuolas de tamaño variable (flechas) que corresponden a silicona, previamente inyectada (H&E ×200). B) Reacción granulomatosa conteniendo estructuras poligonales de hidrogel de metacrilato acrílico (H&E ×400). C) Reacción al compuesto de ácido hialurónico y dextranos (H&E ×400). D) Esferas de polimetilmetacrilato en colágeno bovino (H&E ×400).
Las reacciones histiocitarias tipo xantelasma en párpado inferior tras inyección local de diferentes tipos de rellenos7 se consideran el resultado de una alteración local en el metabolismo de lipoproteínas de baja densidad, más que una reacción directa al material de relleno.
Granulomas por productos no relacionados con la estéticaDefinición: Cualquier material introducido en el cuerpo es capaz de actuar como «cuerpo extraño», siendo detectado por el sistema inmune y provocando una respuesta inflamatoria contra él. Dentro de estos materiales encontramos materiales biológicos (como la queratina del cabello y de la uña, espinas de cactus y otras plantas, así como restos de medusas, corales o erizos de mar), pero también materiales inorgánicos (como tintas de tatuajes, material de sutura, u otros compuestos como la sílice o el talco)8.
Clínica: Existe una gran variedad de presentaciones clínicas por implantación de cuerpo extraño a nivel cutáneo, dependiendo, además, de si nos encontramos ante la fase de respuesta inflamatoria aguda o crónica. En la mayoría de los casos, aparecen pápulas, nódulos o placas de color rojo a marrón, ulcerados o no, que evolucionan hacia el endurecimiento y la fibrosis. También pueden aparecer lesiones de seudofoliculitis (por reacción contra el pelo de la barba o contra el pelo rasurado), abscesos, trayectos fistulosos o incluso lesiones granulomatosas a tipo sarcoidosis (especialmente en las tintas de tatuajes)8.
Histopatología: Tras la reacción inflamatoria aguda neutrofílica desencadenada por la entrada del cuerpo extraño en el organismo, se desarrolla una reacción inflamatoria crónica. Aunque se han descrito diferentes patrones de reacciones tisulares crónicas contra cuerpos extraños (como liquenoides, eccematosas o seudolinfomatosas), la forma de presentación más frecuente es la de tipo granulomatoso8.
Dicho infiltrado granulomatoso (figs. 7A y B) contiene habitualmente macrófagos, que tienden a aumentar de tamaño y formar histiocitos epitelioides o a fusionarse para formar células gigantes multinucleadas tipo cuerpo extraño (figs. 7C y D). Habitualmente aparecen también linfocitos T y fibroblastos8. También se han descrito granulomas de tipo alérgico, con histiocitos epitelioides y una menor densidad de células gigantes multinucleadas tipo Langhans9.
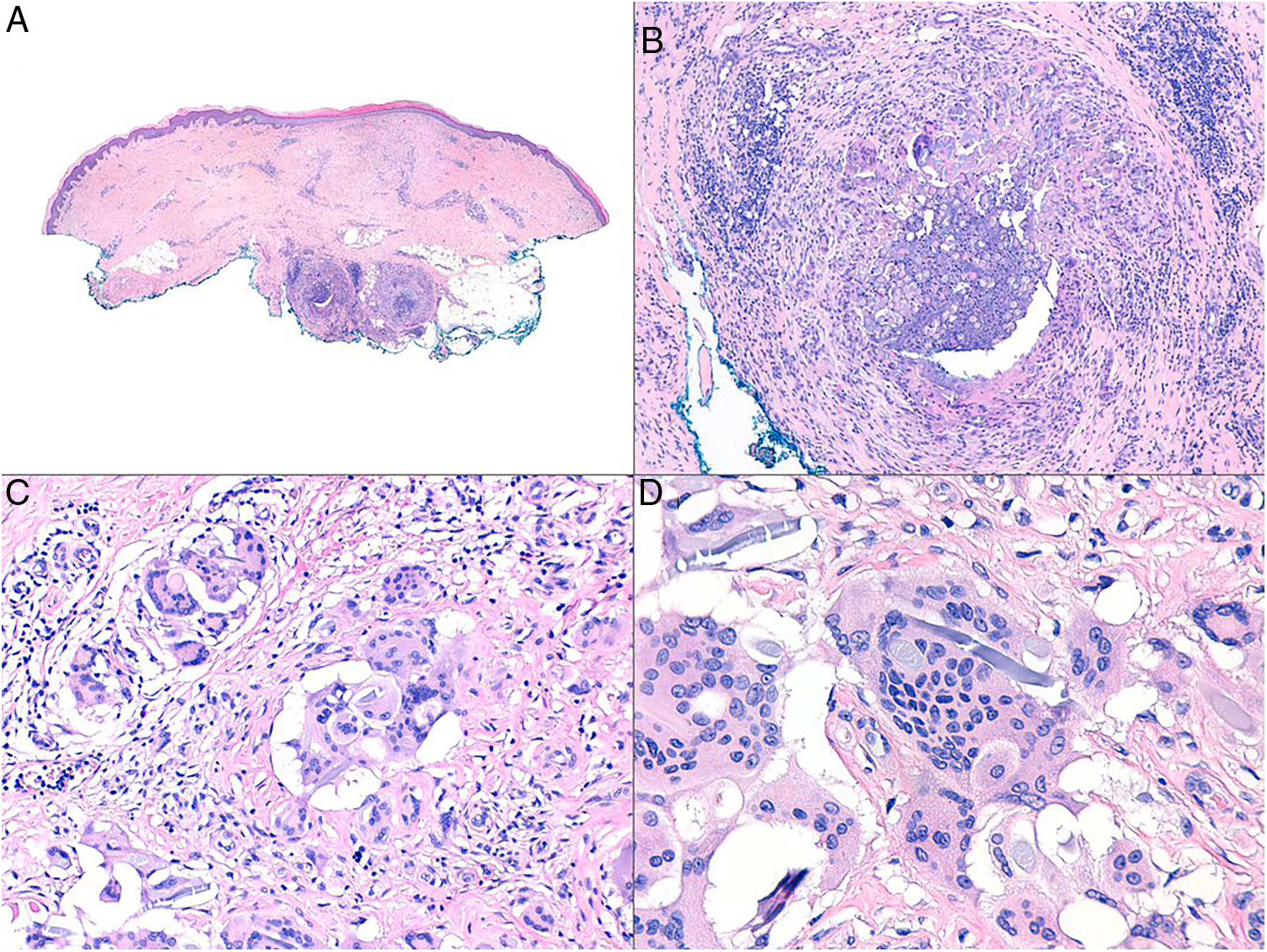
Reacción granulomatosa por material de sutura. A) Panorámica de pieza quirúrgica que muestra 2 granulomas en profundidad (H&E ×40). B) Detalle que muestra un infiltrado inflamatorio mixto, con linfocitos, macrófagos, histiocitos epitelioides y algunas células gigantes multinucleadas a cuerpo extraño, en torno a filamentos de material de sutura (H&E ×200). C) En ocasiones, la célula mayoritaria es la célula gigante multinucleada tipo cuerpo extraño (H&E ×400). D) Células gigante multinucleada digiriendo fibra de material de sutura (H&E ×1.000).
El examen histopatológico puede también confirmar la naturaleza de los cuerpos extraños. La microscopía con luz polarizada pone de manifiesto la birrefringencia positiva de algunos materiales como la sílice, el talco, la queratina, las espinas vegetales, las suturas o las partes de artrópodos. La tinción de PAS es positiva para las astillas de madera, las espinas vegetales y el almidón8.
Granulomas por aluminioDefinición: La administración de vacunas que contienen como adsorbente antigénico sales de aluminio puede producir reacciones locales persistentes en forma de nódulos pruriginosos en el punto de inyección. Se trata de una entidad poco frecuente, con una incidencia calculada en estudios prospectivos del 0,83% de los niños vacunados, posiblemente algo superior en el caso de adultos que realizan tratamientos de desensibilización alergénica10. Estas reacciones locales persistentes se asocian a dermatitis de contacto al hidróxido de aluminio en más del 80% de los pacientes11.
Clínica: Las lesiones consisten en nódulos pruriginosos de localización subcutánea, frecuentemente con cambios epidérmicos secundarios al rascado tales como hipertricosis, liquenificación, escoriaciones y eritema12 (fig. 8A). Las lesiones aparecen varias semanas o meses después de la administración de la vacuna, y pueden persistir a lo largo de los años si no son tratadas.
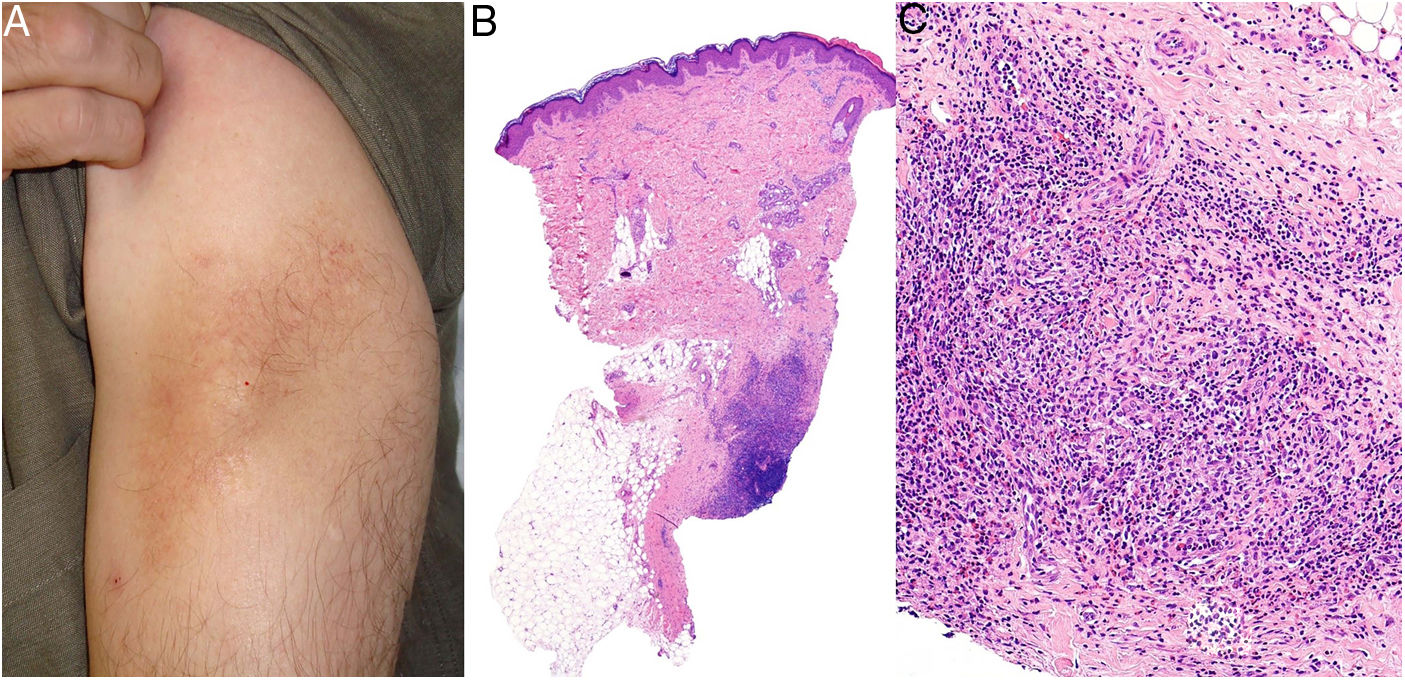
Histopatología: Dada la administración subcutánea de la vacuna, los hallazgos histopatológicos principales se localizan en el tejido celular subcutáneo (fig. 8B). Se han descrito 3 patrones histopatológicos en los nódulos pruriginosos posvacunales por aluminio13. El más frecuente es el de una paniculitis lobulillar, con presencia de linfocitos, plasmocitos, histiocitos y eosinófilos. El segundo en frecuencia es el de un seudolinfoma tipo B, con folículos linfoides con centros germinales bien desarrollado que aparecen separados entre sí por infiltrado histiocitario de apariencia granulomatosa. El tercer patrón es el de un granuloma necrosante en el tejido celular subcutáneo, con presencia de linfocitos, plasmocitos y eosinófilos (fig. 8C). La clave para el diagnóstico en todos estos patrones la otorga la presencia de histiocitos de citoplasma amplio, violáceo y granular (fig. 9A), que se forma como consecuencia del depósito intracelular de sales de aluminio, y que se puede confirmar mediante técnicas de microscopía electrónica (fig. 9B), aunque recientemente se ha demostrado que la hibridación in situ EBER tiñe positivamente los gránulos, permitiendo el diagnóstico diferencial con otro tipo de granulomas14.
Necrobiosis lipoidea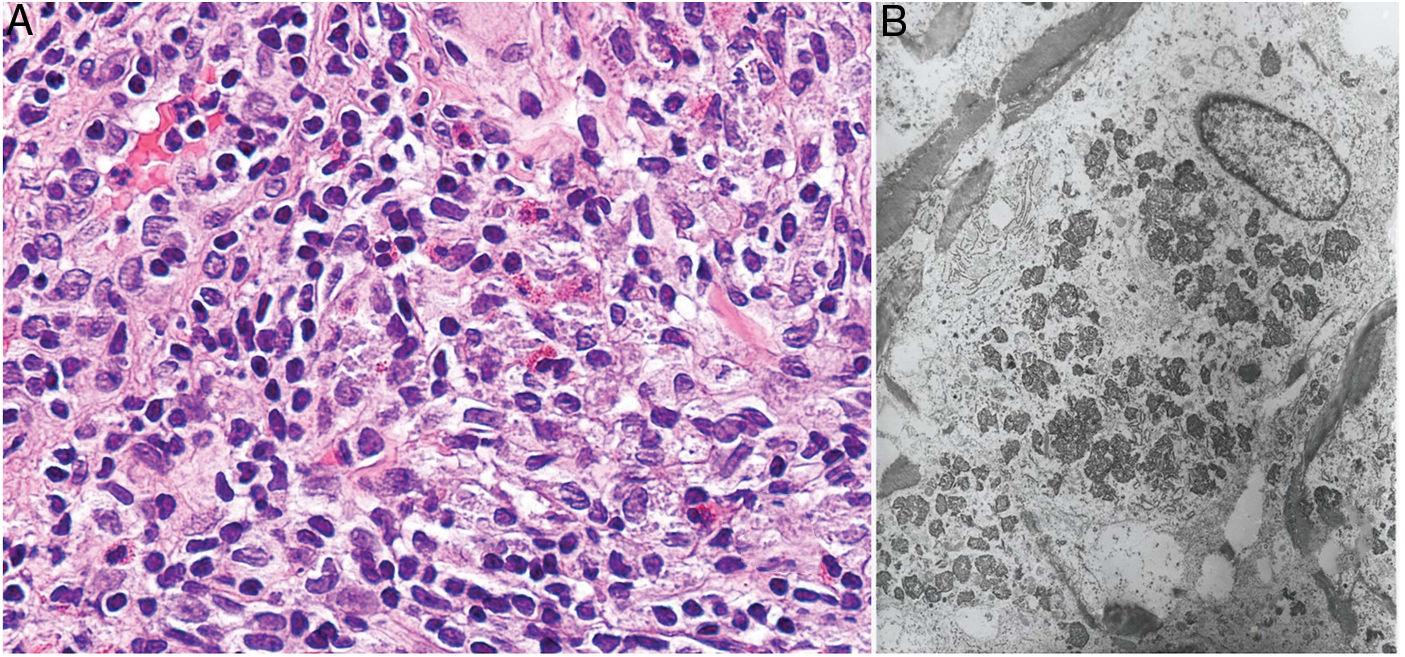
Definición: La necrobiosis lipoidea (NL) es una enfermedad granulomatosa crónica poco frecuente, clásicamente asociada a la diabetes mellitus (DM). A pesar de haber sido asociada a la DM desde su primera descripción, se estima que menos del 1% de los pacientes diabéticos desarrollan NL, y el porcentaje de pacientes con DM en las series de NL disponibles en la literatura es muy variable15–18. Su origen y patogénesis no han sido aún establecidos.
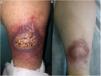
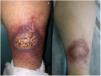
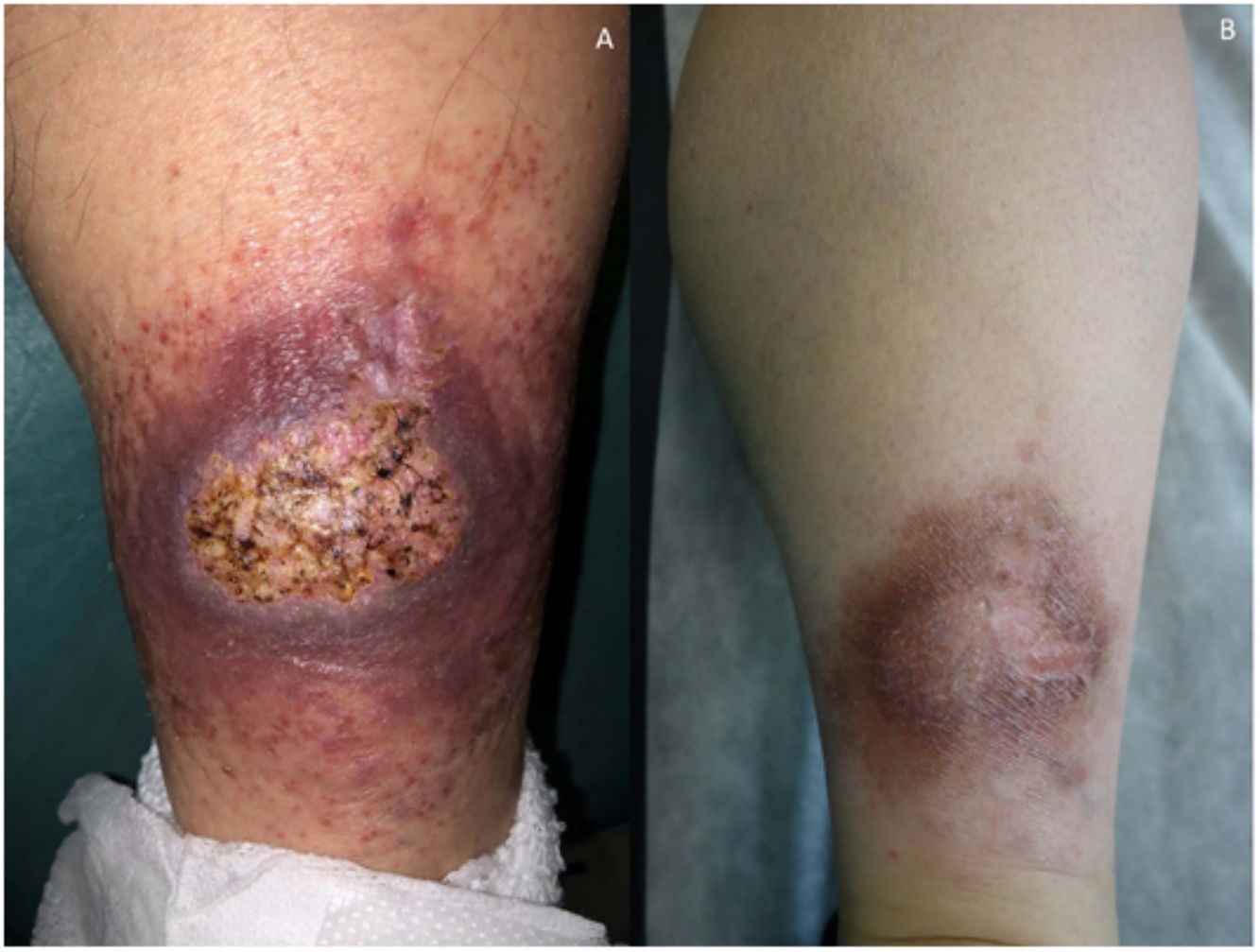
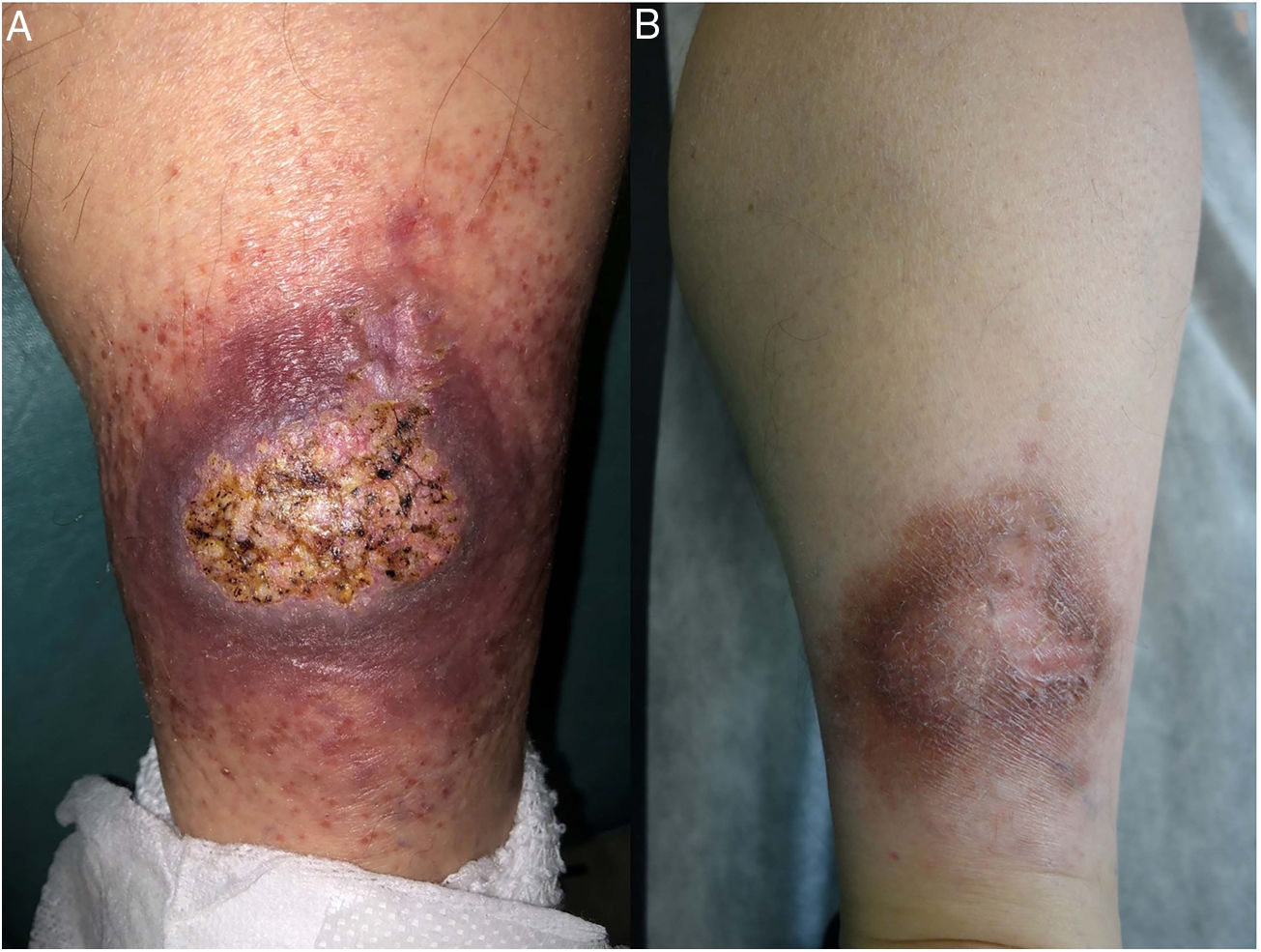
Clínica: La NL afecta típicamente a adultos de edad media, con un predominio femenino15–17. Clínicamente, se caracteriza por lesiones papulares y nodulares violáceas o eritematosas, que evolucionan progresivamente a placas coalescentes, de coloración amarillenta o marronácea. El centro de las placas suele ser atrófico, presenta teleangiectasias, y es frecuente la ulceración secundaria15,17 (fig. 10A). La localización más frecuente es la región pretibial, pero también las lesiones pueden aparecer en áreas tales como las extremidades superiores, la cara y el cuero cabelludo15,17,18. Aunque suelen ser asintomáticas, las lesiones pueden generar prurito o disestesias, o incluso ser dolorosas18. Las lesiones de NL pueden ser resistentes a múltiples tratamientos18,19 y, al menos en algunos casos, las lesiones podrían remitir parcial o totalmente con un mejor control de la DM16.
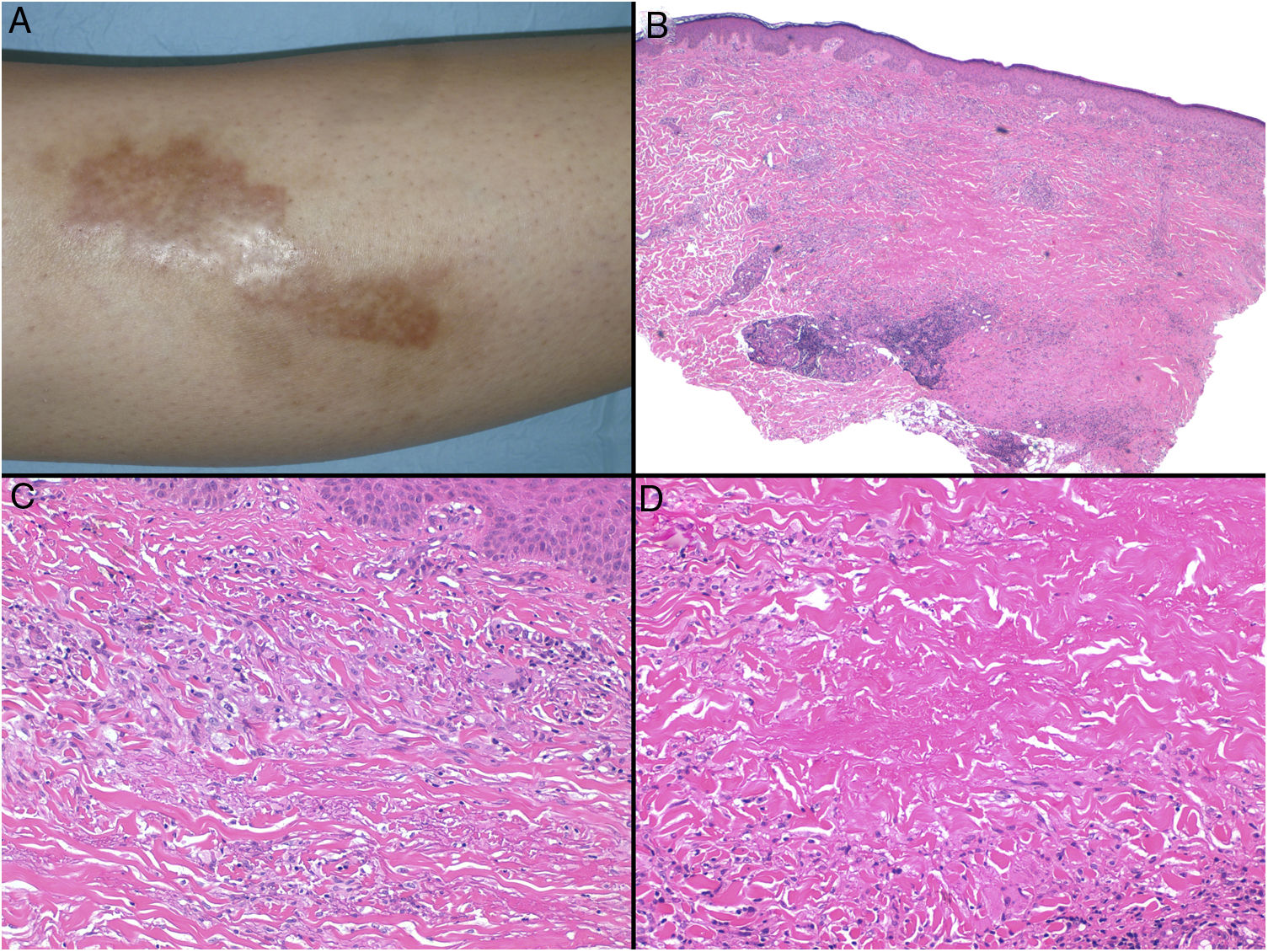
Necrobiosis lipoidea. A) Placa infiltrada de tonalidad eritemato-amarillenta y aspecto céreo, localizada en región pretibial. El centro de la placa, más claro, tiene un aspecto atrófico. B) En la visión panorámica se aprecia un infiltrado inflamatorio focal que parece dispuesto en capas más o menos paralelas a la epidermis, así como áreas de engrosamiento del colágeno, que se muestra intensamente eosinófilo. En la parte inferior de la biopsia, en dermis profunda, se observan agregados linfoides de distribución perianexial (H&E ×40). C) A mayores aumentos se observa un infiltrado inflamatorio granulomatoso intersticial, dispuesto entre haces de colágeno engrosado y necrobiótico. Pueden verse algunos histiocitos multinucleados (H&E ×200). D) En otra zona de la biopsia, a grandes aumentos, observamos un granuloma de disposición en empalizada, centrado por un área de marcada necrobiosis del colágeno (H&E ×400).
Histopatología: Los hallazgos histopatológicos típicos se observan en el borde inflamatorio, activo, de la lesión. Histológicamente, se caracteriza por un infiltrado granulomatoso intersticial centrado en la dermis reticular, que frecuentemente afecta también al tejido celular subcutáneo en forma de paniculitis septal (fig. 10B). Característicamente, este infiltrado granulomatoso se dispone paralelamente a la epidermis, de modo que se alternan capas de infiltrado histiocitario con otras de necrobiosis del colágeno15,16,20 (fig. 10C). Esta afectación «en capas» es más frecuente en la NL asociada a la DM. También es posible observar áreas de granulomas en empalizada, con un centro compuesto por colágeno necrobiótico, y células gigantes multinucleadas15,19,20 (fig. 10D). Generalmente, el depósito de mucina es ausente o escaso. Puede haber agregados linfoides y un infiltrado perivascular superficial en la dermis adyacente, y los vasos suelen tener una pared engrosada y una luz ligeramente reducida, a expensas de un material PAS positivo (principalmente en la NL asociada a la DM)15,16,20,21. Paralelamente, puede demostrarse la presencia extracelular de depósitos lipídicos mediante la tinción de adipofilina21. Sus principales diagnósticos diferenciales histológicos incluyen otros trastornos granulomatosos, y otras formas de paniculitis septal.
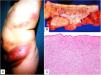
Xantogranuloma necrobióticoDefinición: El xantogranuloma necrobiótico (XGN) es una histiocitosis no-Langerhans, asociada frecuentemente a paraproteinemia en el contexto de discrasias de células plasmáticas u otros desórdenes linfoproliferativos22.
Clínica: La enfermedad se presenta a una edad media de 61 años, con lesiones frecuentemente múltiples, de tipo pápulas, placas y en menor proporción nódulos, de coloración amarillento-anaranjada, rojiza o violácea (fig. 11)23–25. La localización más frecuente es la cara, especialmente la región periorbitaria, seguida de la afectación en extremidades y tronco. El compromiso extracutáneo está descrito en un porcentaje cercano al 30%, siendo el ojo el órgano más frecuentemente afectado25.
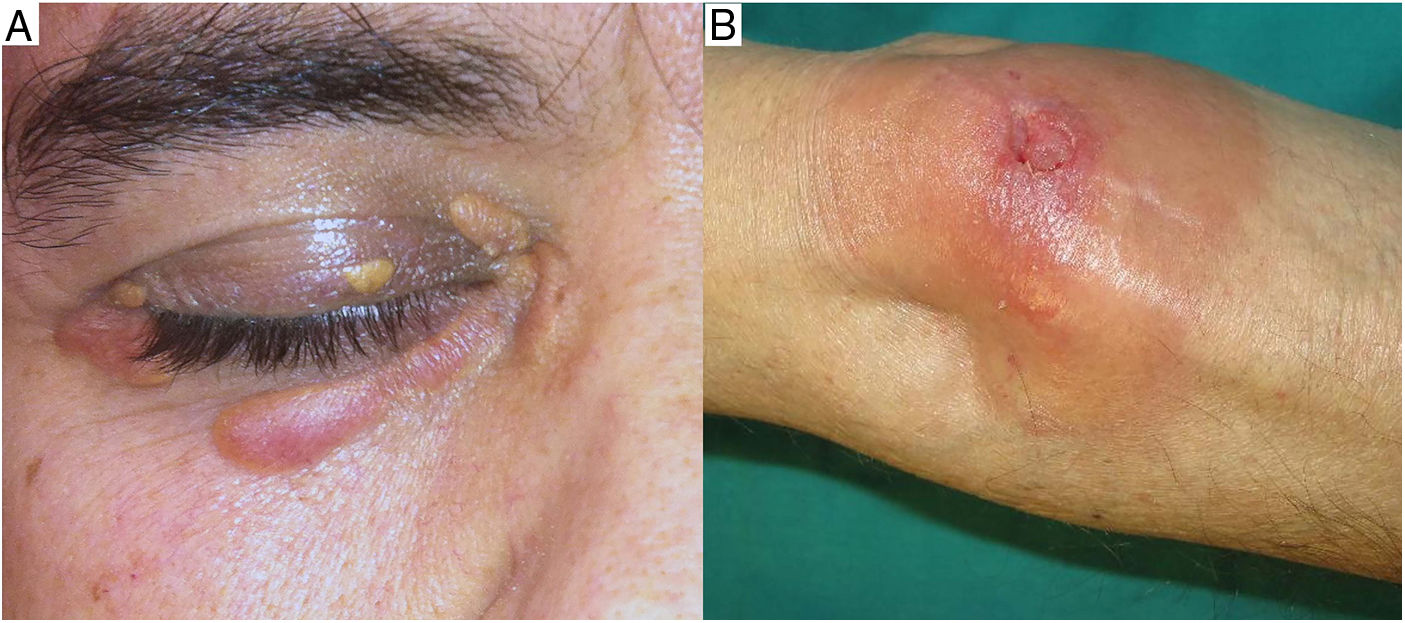
Como hallazgos analíticos destacan las alteraciones del perfil lipídico, en especial la elevación del colesterol total y las proteínas de baja densidad; los niveles bajos de C4 y la presencia de crioglobulinas24,25.
Histopatología: Las lesiones muestran hallazgos similares a la necrobiosis lipoidica (NL), con presencia de granulomas «rojos» asociados a necrobiosis eosinofílica, y un infiltrado histiocitario de distribución en banda que compromete dermis y tejido celular subcutáneo. El infiltrado histiocitario muestra células espumosas, células gigantes de tipo Touton y/o de tipo reacción frente a cuerpo extraño, asociados a nódulos de infiltrado linfocitario con numerosas células plasmáticas, sin restricción de cadenas ligeras26. Adicionalmente, hay cristales de colesterol extracelulares (fig. 12)27. Un estudio realizado usando inmunohistoquímica para adipofilina, ha demostrado positividad extra e intracelular, a diferencia de la NL que muestra una tinción circunscrita a las áreas de necrobiosis28.
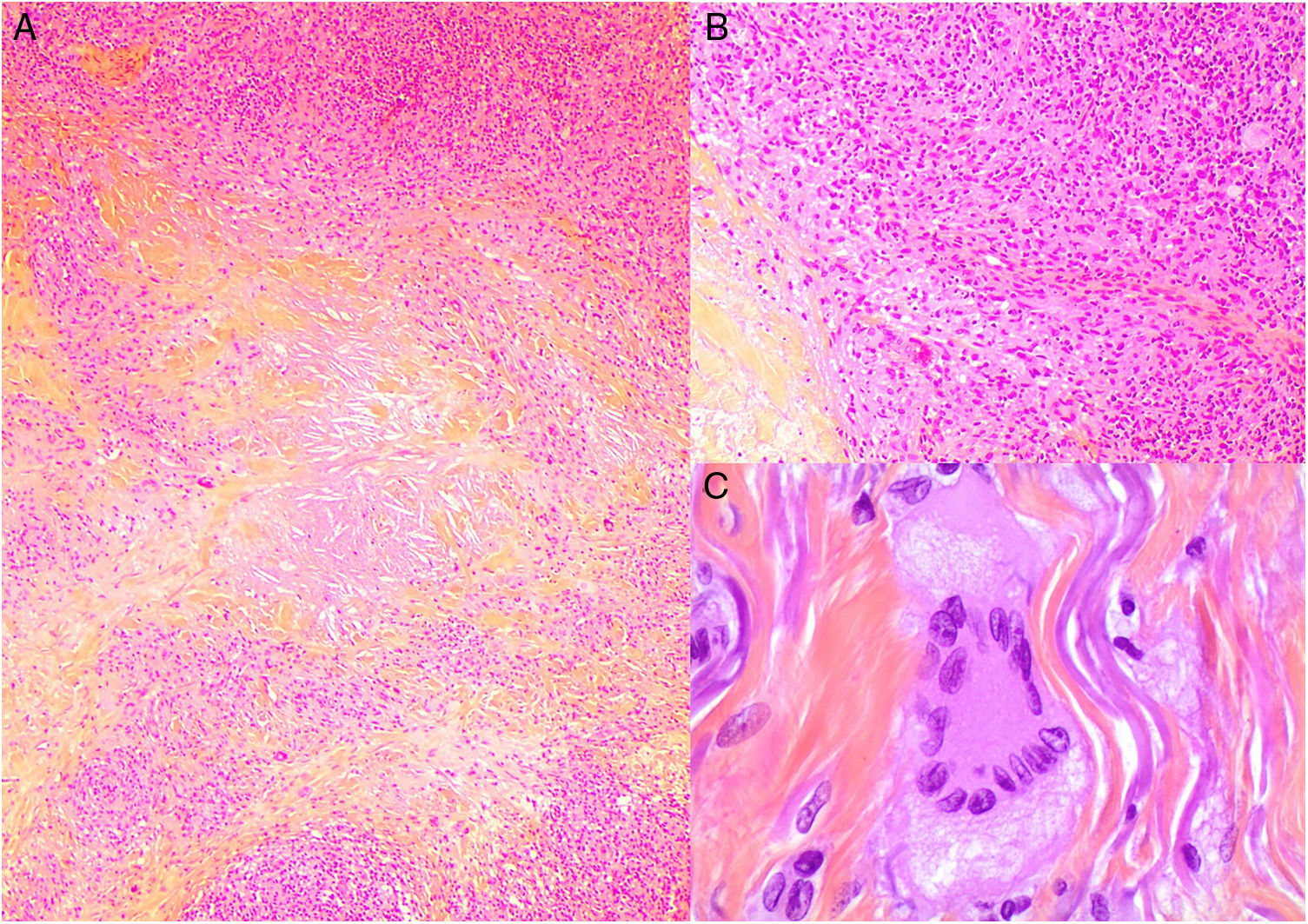
Histología del xantogranuloma necrobiótico. A) Infiltrado granulomatoso rodeando una zona central de necrobiosis eosinofílica con cristales de colesterol asociados (H&E ×40). B) El infiltrado histiocitario se acompaña de nódulos linfoides y células plasmáticas (H&E ×100). C) Detalle de una célula de Touton (H&E ×400).
Recientemente se han propuesto unos criterios para el diagnóstico del XGN25 (tabla 1). Se requieren ambos criterios mayores y uno menor para el diagnóstico, aplicables en ausencia de cuerpo extraño, infección u otras causas identificables.
Tabla de criterios diagnósticos propuestos para el diagnóstico de xantogranuloma necrobiótico. Se requieren ambos criterios mayores y uno menor
| Criterios mayores |
| 1. Pápulas, placas y/o nódulos de coloración amarillenta o anaranjada |
| 2. Histopatología con granulomas en empalizada acompañados de infiltrado linfo-plasmocitario y necrobiosis. De forma variable puede haber cristales de colesterol y/o células gigantes (Touton o tipo cuerpo extraño) |
| Criterios menores |
| 1. Paraproteinemia, más frecuentemente IgG-kappa, discrasia de células plasmáticas y/u otro desorden linfoproliferativo |
| 2. Distribución periorbitaria de las lesiones |
Definición: La enfermedad inflamatoria intestinal (EII) incluye 2 entidades principales: la enfermedad de Crohn (EC) y la colitis ulcerosa (CU). Aunque, sus manifestaciones son principalmente gastrointestinales, las manifestaciones extra-intestinales (MEI) aparecen hasta en el 40% de los casos. Además, la prevalencia de MEI es mayor en la EC en comparación con la CU29. Casi cualquier órgano puede verse afectado en las EII, incluidos los ojos, la piel, los pulmones, los riñones, el hígado, el sistema inmunológico, el hematológico y el cardiovascular29. La piel es uno de los órganos más comúnmente afectados, y las manifestaciones cutáneas pueden estar presentes en el 10-22% de estos pacientes29,30.
Clínica: Se han descrito 5 categorías para clasificar las manifestaciones mucocutáneas asociadas con la EII según su asociación fisiopatológica con la enfermedad subyacente:
- 1.
Manifestaciones específicas con las mismas características histológicas de la EII.
- 2.
Trastornos mucocutáneos asociados con la EII: Las lesiones mucocutáneas más comunes asociadas con las EII son el eritema nodoso (EN), el pioderma gangrenoso (PG) y la estomatitis aftosa31.
- 3.
Manifestaciones reactivas de las EII debido a mecanismos inmunológicos desencadenados por antígenos compartidos por las bacterias intestinales y la piel.
- 4.
Condiciones mucocutáneas secundarias al tratamiento de las EII.
- 5.
Manifestaciones por malabsorción nutricional.
La aparición de granulomas cutáneos no caseificantes específicos en sitios distantes del tracto gastrointestinal se conoce como enfermedad de Crohn metastásica (ECM)29. Las áreas más frecuentemente afectadas son las extremidades inferiores y las áreas intertriginosas, aunque puede verse en cualquier localización (fig. 13). Las manifestaciones clínicas de la ECM consisten en placas eritematosas, nódulos, abscesos, fístulas y úlceras. No hay una correlación clara entre el desarrollo de ECM y la gravedad de la EC subyacente.
Histopatología: La imagen histológica de la ECM puede ser similar al de la EC del intestino: granulomas estériles, no caseificantes, con células gigantes de cuerpo extraño o células de Langhans, histiocitos epitelioides y células plasmáticas (figs. 14A y B)32. Ocasionalmente, los granulomas se ven alrededor de vasos sanguíneos de apariencia normal. Frecuentemente, se asocia a infiltrado linfocitario en dermis reticular que puede extenderse al tejido subcutáneo. Otras características menos comunes incluyen degeneración de colágeno, infiltrado eosinofílico, ulceración, edema dérmico y dermatitis liquenoide.
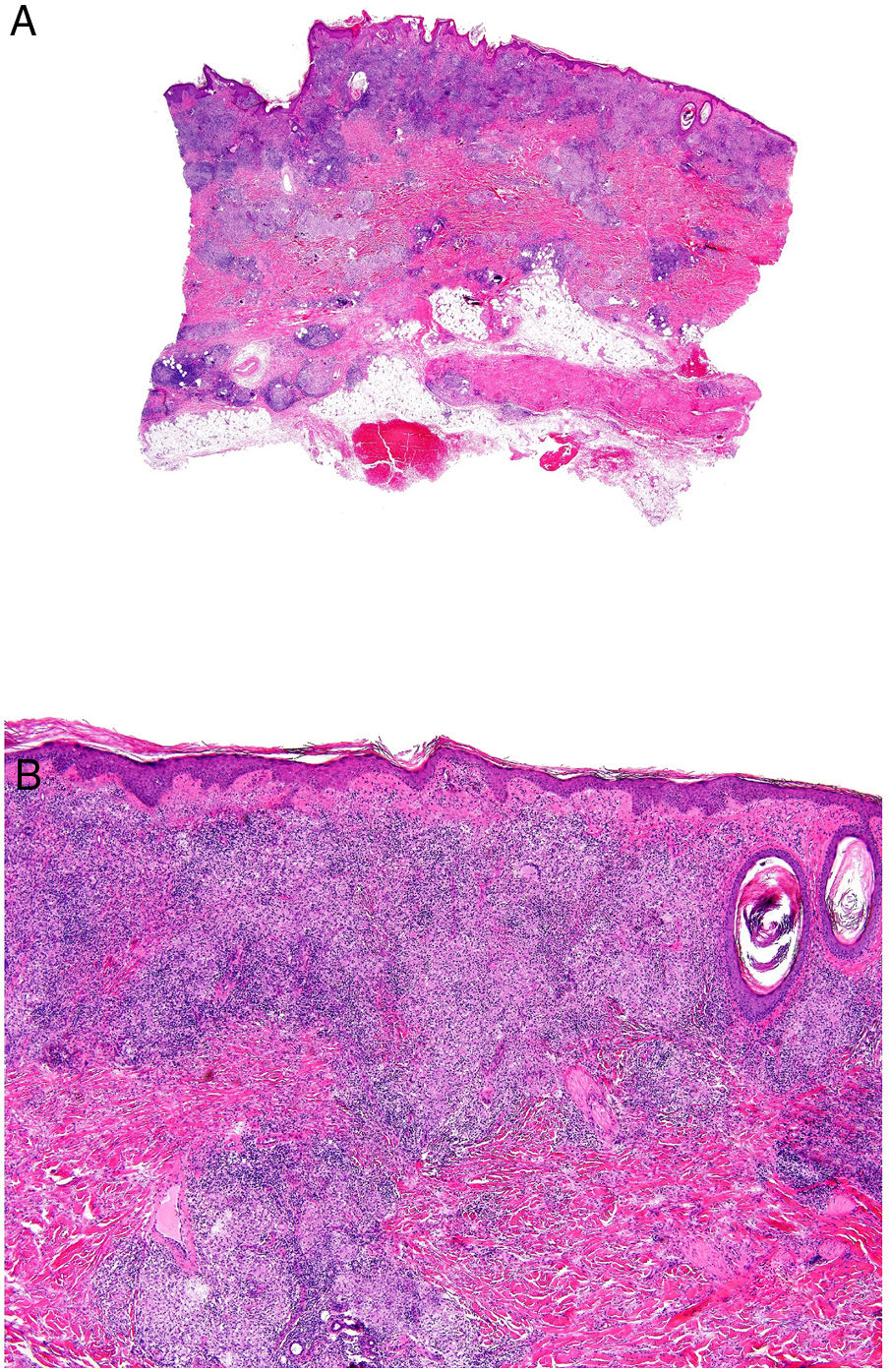
Enfermedad de Crohn. Dermatitis granulomatosa con múltiples granulomas en la dermis reticular, respetando la epidermis y la dermis papilar (A: H&E ×20). Los granulomas son similares a los que se observan en el tracto digestivo: no caseificantes, con células gigantes multinucleadas, y pueden tener un denso infiltrado linfocitario (B: H&E ×40).
Definición: Subtipo muy infrecuente de micosis fungoide (MF) de curso indolente, con una supervivencia de casi el 100% a los 5 años, cuyo diagnóstico se basa en datos clínico-patológicos33.
Clínica: Se presenta con mayor frecuencia en pacientes jóvenes, y cursa con placas y pápulas eritematosas, de aspecto «penduloso», flácido, en las flexuras corporales (predilección por axilas e ingles), de lento crecimiento durante años (fig. 15A). Puede coexistir en su evolución con un proceso hematológico sistémico (linfoma de Hodgkin, leucemia mieloide aguda, histiocitosis de células de Langerhans).
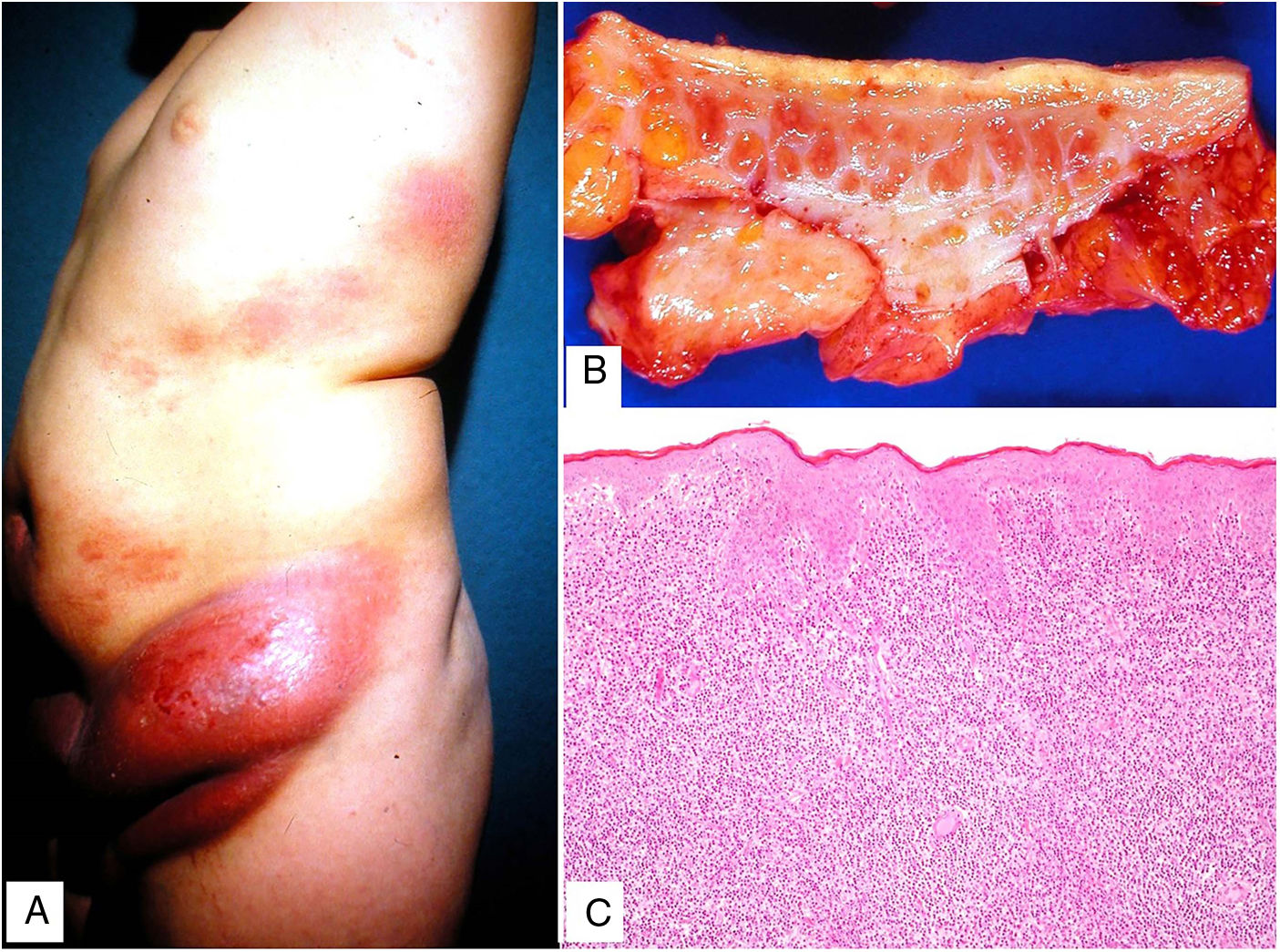
Histopatología: Infiltrado dérmico difuso de linfocitos atípicos de pequeño tamaño (inmunofenotipo T helper: CD3+, CD4+ y CD8−), que puede extenderse a hipodermis (figs. 15B y C), con numerosas células gigantes multinucleadas (figs. 16A y B) que muestran elasto- y linfo-fagocitosis (figs. 16C y D). En fases iniciales, la histología puede ser similar a una micosis fungoide convencional. Con PCR se demuestra reordenamiento del receptor de célula T (TCR)34.
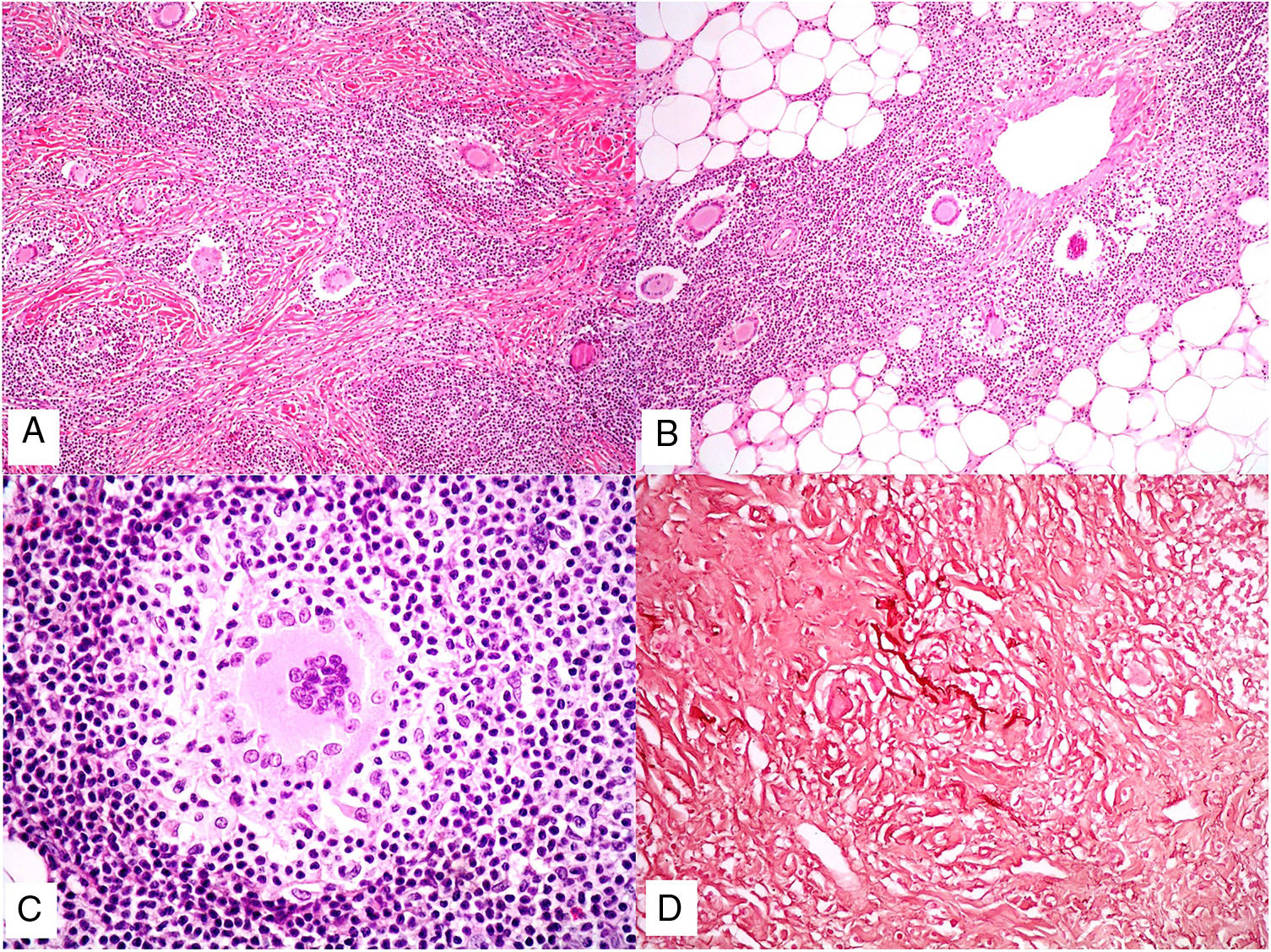
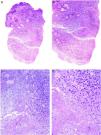
Piel laxa granulomatosa. A) Infiltración linfoide en dermis con numerosas células gigantes multinucleadas salpicadas (H&E ×40). B) Infiltración linfoide en septos de la hipodermis con células gigantes multinucleadas (H&E ×40). C) Detalle de una célula gigante con linfofagocitosis (H&E ×200). D) Fragmentación y pérdida de fibras elásticas (orceína ×100).
No debe de confundirse con la MF granulomatosa (células gigantes con menor número de núcleos y sin linfofagocitosis evidente). Pueden observarse histiocitos multinucleados de modo focal en la MF en fase tumoral, en ocasiones por efecto del tratamiento o bien como consecuencia de la rotura de folículos pilosebáceos. En todo caso, siempre es necesario una adecuada correlación clínico-patológica33,35.
Micosis fungoide granulomatosaDefinición: Variante exclusivamente histopatológica de micosis fungoide, caracterizada por la presencia de granulomas36.
Clínica: Los casos descritos han consistido en manchas, placas o tumores, sin que el aspecto clínico permitiera sospechar la presencia del infiltrado granulomatoso (fig. 17A). Aunque se atribuyó inicialmente a la inflamación granulomatosa un efecto protector, el pronóstico de las micosis fungoides con granulomas parece ser peor que el de las variantes clínicas sin este componente histológico.
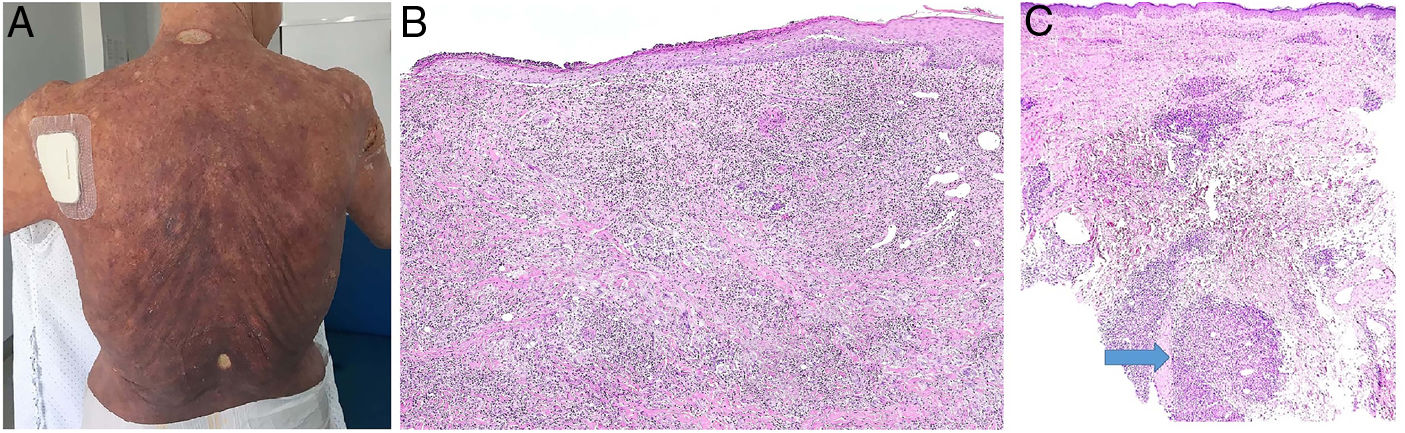
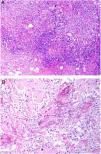
A) Micosis fungoide granulomatosa: Afectación extensa de la espalda por micosis fungoide (histopatológicamente granulomatosa) en una paciente de 77 años con lesiones diseminadas de larga evolución, mostrando placas y nódulos tumorales focalmente ulcerados (foto cortesía del Dr. César Pérez-Vega). B) Micosis fungoide granulomatosa: Además de afectación de la epidermis por linfocitos atípicos, infiltración transmural de la dermis que incluye células gigantes y granulomas de variable textura (H&E ×20). C) Linfoma T no micosis fungoides. Infiltración dérmica intersticial por linfoma T periférico con formación de un nódulo dérmico profundo en el que se localizan granulomas epitelioides confluyentes de tipo sarcoideo (flecha) con relación a células del linfoma (H&E ×10).
Histopatología: En los casos publicados, la cuantía y las características de los granulomas es muy variable: desde aisladas células gigantes multinucleadas hasta granulomas epitelioides compactos, pasando por infiltrados granulomatosos mal definidos. Su distribución suele ser amplia, ocupando todo el espesor de la dermis (fig. 17B); excepcionalmente se ha descrito un patrón exclusivamente superficial37. La conjunción de afectación de tejido subcutáneo y la clínica de pliegues péndulos es propia de la piel laxa granulomatosa, una de las 3 variantes reconocidas por la clasificación de la OMS, como se explica anteriormente; la distinción respecto de la micosis fungoide granulomatosa requiere correlación clinicopatológica. Se ha descrito la presencia en esta última de grupos de células con expresión de CD12338.
Otros linfomasDefinición: El patrón de reacción granulomatosa es un hallazgo poco frecuente (1,6-1,8%)39, pero bien conocido en asociación a linfomas tanto de estirpe T como B, de alto o bajo gado, primarios cutáneos o sistémicos con afectación secundaria de la piel.
Clínica: La presentación clínica es poco específica. Se han descrito: rash, placas eritematosas, placas arciformes40 o anulares40,41 no simétricas42 y nódulos43.
Histopatología: El patrón de reacción granulomatosa puede observarse en contexto de linfomas B y T, de alto y bajo grado, primarios cutáneos y sistémicos afectando la piel. La histología de la población linfocitaria es, por tanto, muy variada y se corresponde con la de la neoplasia de base. En general, en linfomas primarios cutáneos se observa una reacción granulomatosa dérmica o dermo-hipodérmica de intensidad variable con relación a la infiltración por el linfoma44 (fig. 17C). En ocasiones, la abundancia de granulomas puede derivar en diagnóstico de dermatitis granulomatosa, y producir retraso en la identificación del linfoma39,45. En la literatura se recogen casos de: linfoma T CD30+ cutáneo, linfoma T tipo paniculítico, linfoma B de la zona marginal primario cutáneo y linfoma folicular39,44.
En linfomas sistémicos con afectación de la piel se reconocen 2 tipos de reacción granulomatosa. En el primer tipo se pueden objetivar células tumorales, y en el segundo se observa reacción granulomatosa, pero no linfoma. Los granulomas pueden ser necrosantes de tipo tuberculoideo, necrobióticos en empalizada o sarcoideos40. No queda claro si los granulomas se producen por la presencia del linfoma o si son una respuesta inmune peculiar del huésped. No se debe olvidar, además, la posibilidad de infecciones oportunistas en estos pacientes, que se deben descartar. Entre los linfomas sistémicos con afectación cutánea destacan: linfoma T periférico, linfoma T angioinmunoblástico, linfoma B difuso de célula grande, linfoma de Hodgkin, leucemia/linfoma T del adulto, síndrome de Sezary y linfoma T rico en histiocitos39,41,43,44.
Se desconoce, por el momento, el significado pronóstico de la presencia de granulomas con relación a los procesos linfoproliferativos primarios o secundarios localizados en la piel45.
Alopecias con respuesta granulomatosaDefinición: Algunas alopecias pueden mostrar diversos tipos de granulomas. En algunos casos, el proceso granulomatoso está directamente relacionado con la etiología de la alopecia, por ejemplo, como en algunas alopecias de origen infeccioso. En otros, el proceso granulomatoso forma parte de los hallazgos de una patogenia incierta o no del todo conocida. Por último, en otros, el granuloma es una respuesta a la salida del tallo piloso y otros productos del folículo por destrucción de este último.
Clínica: Los procesos granulomatosos se saldan en su mayoría con la destrucción de las estructuras adyacentes a la respuesta granulomatosa y, por lo tanto, con la lesión irreversible de la protuberancia del folículo piloso. Esto conlleva la desaparición irreparable del folículo y, en consecuencia, una alopecia cicatricial irreversible. La clínica adicional de base, dependerá del tipo de alopecia del que se trate.
La alopecia central centrífuga cicatricial (ACCC) muestra una alopecia centrada en el vertex de intensidad variable y con un frente inflamatorio centrífugo de avance. Las alopecias neutrofílicas (foliculitis decalvante, celulitis disecante y acné keloidalis) muestran áreas induradas y edematosas que progresan a la induración y a la cicatrización con pérdida irreversible del cabello. Las áreas afectas varían según la entidad: occipital y vértex en la foliculitis decalvante; vértex y coronilla en la celulitis disecante; nuca en el acné queloideo.
Histopatología: En la ACCC, el adelgazamiento asimétrico de la parte alta del folículo juega un papel fundamental. Ello conlleva la rotura de la pared del folículo con la extrusión del tallo a la dermis y la inducción de una respuesta granulomatosa a cuerpo extraño (fig. 18A).
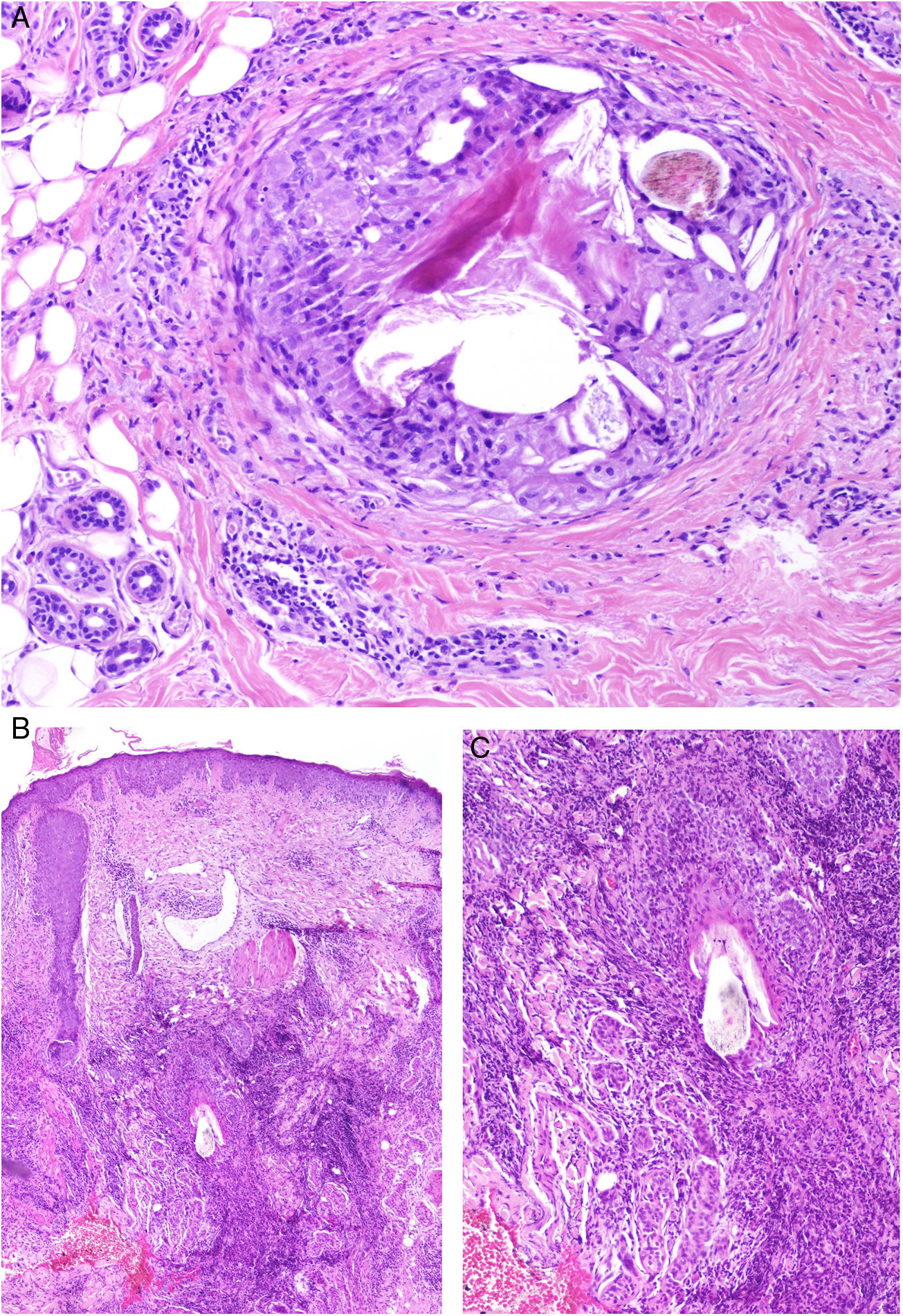
A) Alopecia central centrífuga cicatricial. Presencia de un granuloma a cuerpo extraño frente a fragmentos de un tallo piloso y láminas de queratina (H&E ×100). B y C) Foliculitis decalvante. En estadios avanzados pueden observarse granulomas a cuerpo extraño frente fragmentos de tallos pilosos (B: H&E ×20 y C: H&E ×100).
En el grupo de alopecias «neutrofílicas» (foliculitis decalvante, celulitis disecante y acné keloidalis), el infiltrado inflamatorio puede ser granulomatoso en estadios avanzados (fig. 18B). Se trata de granulomas a cuerpo extraño, generalmente en respuesta a fragmentos de tallo piloso (fig. 18C). A veces los granulomas son de tipo abscesificado, pero la búsqueda de hongos o micobacterias resulta infructuosa.
Vasculitis asociadas a anticuerpos citoplasmáticos antineutrófilosDefinición: Las vasculitis asociadas a anticuerpos citoplasmáticos antineutrófilos (ANCA) son un grupo heterogéneo de enfermedades autoinmunes raras que provocan una inflamación de los vasos sanguíneos con diversas manifestaciones. Incluye 3 cuadros principales: la granulomatosis con poliangeítis (GP), anteriormente conocida como granulomatosis de Wegener, la granulomatosis eosinofílica con poliangeítis (GEP), anteriormente conocida como síndrome de Churg-Strauss y la poliangeítis microscópica (PAM). Otras enfermedades asociadas a ANCA son la vasculitis inducida por fármacos y la vasculitis renal limitada46.
Clínica:
- 1.
La GP suele presentarse en la cuarta y quinta décadas de la vida, con síntomas relacionados con el tracto respiratorio superior, como rinorrea persistente, dolor sinusal, tos y hemoptisis. Las manifestaciones cutáneas ocurren entre el 30 y el 50% de los casos y, en ocasiones, pueden ser el síntoma inicial. Consisten en lesiones papulonecróticas distribuidas simétricamente sobre los codos, las rodillas y, a veces, las nalgas47.
- 2.
La GEP afecta a individuos de mediana edad con predominio en los varones. Los criterios del American College of Rheumatology requieren 4 de los siguientes 6 hallazgos para el diagnóstico de GEP: asma, eosinofilia mayor al 10%, sinusitis paranasal, infiltración pulmonar, prueba histológica de vasculitis y mononeuritis múltiple48. La afectación cutánea puede ser en forma de lesiones hemorrágicas (que varían desde petequias hasta púrpura palpable, equimosis extensas, a veces acompañadas de úlceras necróticas) y nódulos cutáneos o subcutáneos.
- 3.
La PAM es una vasculitis sistémica de vasos pequeños que se asocia típicamente con glomerulonefritis necrosante focal con semilunas y autoanticuerpos citoplasmáticos antineutrófilos de mieloperoxidasa sérica (p-ANCA). La mayoría de los pacientes son varones de más de 50 años. La característica clínica más común es la nefropatía, que se manifiesta como microhematuria, proteinuria o insuficiencia renal oligúrica aguda. Se acompaña de síntomas prodrómicos que incluyen fiebre, mialgias, artralgias y dolor de garganta. Al menos entre el 30 y el 40% de los pacientes con PAM presentan cambios en la piel que incluyen máculas eritematosas, livedo reticularis, púrpura palpable, hemorragias en astilla y ulceraciones46.
Histopatología:
- 1.
En la GP, la mayoría de las biopsias muestran hallazgos inespecíficos, con infiltrados linfocitarios perivasculares. Sin embargo, entre un 25 y un 50% de los pacientes presentan vasculitis leucocitoclástica de vasos pequeños e inflamación granulomatosa necrosante49. Los cuadros granulomatosos contienen histiocitos, linfocitos y células gigantes con necrosis supurativa formando los denominados granulomas en empalizada «azules»: granulomas que rodean restos de colágeno necrobiótico y detritos necróticos basófilos mezclados con neutrófilos50 (fig. 19A). Además de estas lesiones, que no son patognomónicas de la GP, puede observarse vasculitis granulomatosa verdadera, inflamación granulomatosa perifolicular acneiforme, cuadros similares al eritema nodoso y dermatosis neutrofílica en empalizada47.
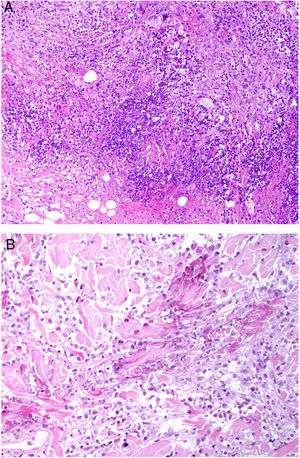 Figura 19.
Figura 19.A) Granulomatosis con poliangeitis. Infiltrado granulomatoso con granulomas mal formados constituidos por histiocitos, linfocitos y células gigantes con necrosis supurativa y restos de colágeno necrobiótico (H&E ×100). B) Granulomatosis eosinofílica con poliangeitis. Granuloma constituido por histiocitos dispuestos radialmente alrededor de fibras de colágeno degeneradas con restos de eosinófilos (H&E ×200).
- 2.
En la GEP, las áreas de hemorragia cutánea suelen mostrar vasculitis leucocitoclastica de vasos pequeños con numerosos eosinófilos51. En algunos casos, la dermis muestra un tipo de granuloma característico constituido por histiocitos y células gigantes multinucleadas dispuestos radialmente alrededor de fibras de colágeno degeneradas con restos de eosinófilos (granulomas «rojos»)52 (fig. 19B). Inicialmente se pensó que estos granulomas eran característicos de la enfermedad, y se denominaron granulomas de Churg-Strauss. Sin embargo, ni están siempre presentes y ni son un requisito para el diagnóstico. Además, también se pueden observar en otros cuadros, como artritis reumatoide, lupus eritematoso, procesos linfoproliferativos, endocarditis bacteriana subaguda, hepatitis crónica activa y EII. Los granulomas extravasculares pueden localizarse en la grasa subcutánea, donde pueden alcanzar un tamaño considerable, dando lugar a la aparición de nódulos.
- 3.
En la PAM se observa una vasculitis leucocitoclástica con afectación de arteriolas, vénulas y capilares. Solo, en ocasiones, se observa una vasculitis necrosante de arterias de tamaño mediano, típica de la panarteritis nodosa clásica. La presencia de componente inflamatorio granulomatoso en las lesiones cutáneas no es lo habitual.
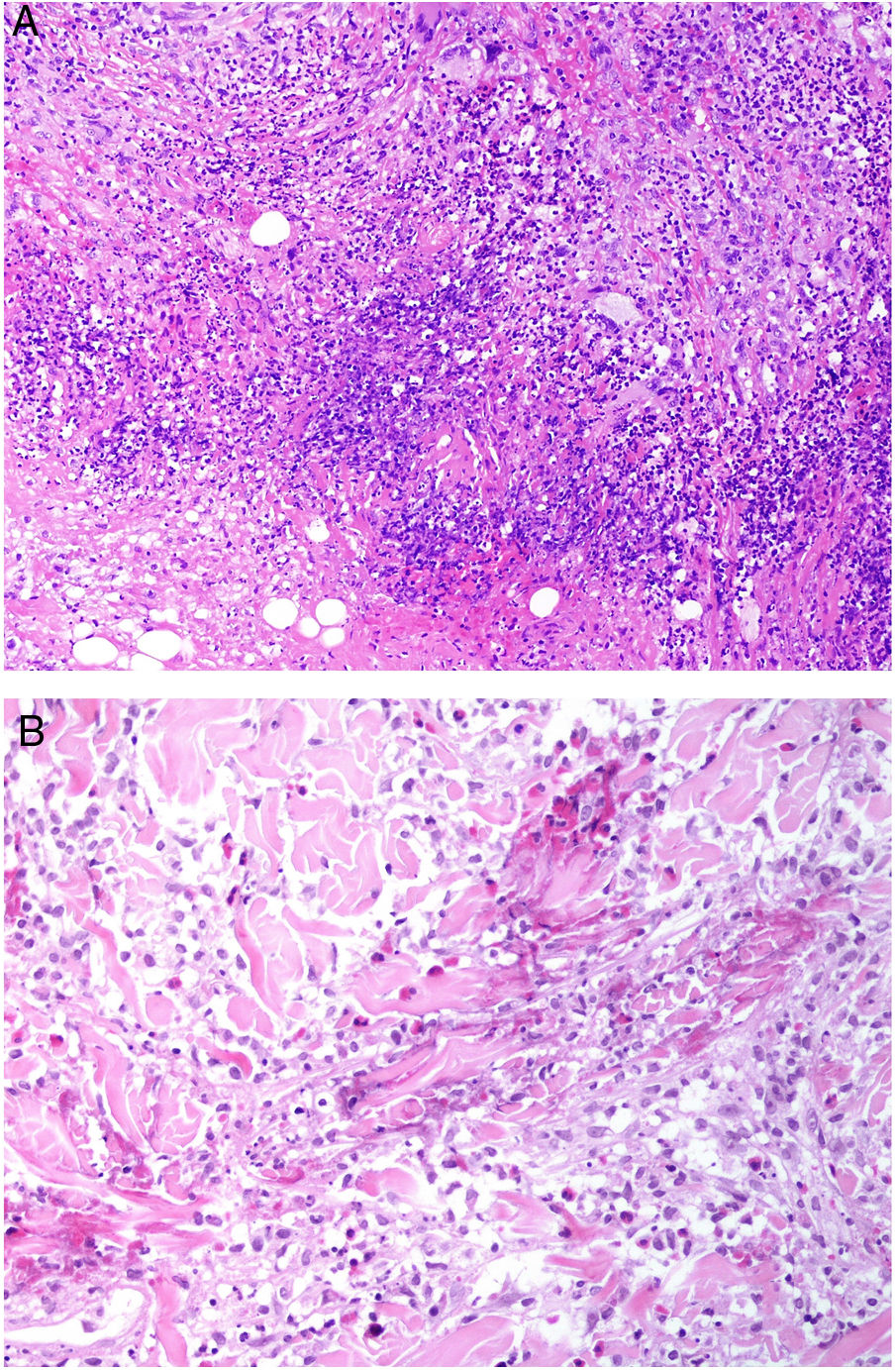
Definición: La fiebre reumática es una enfermedad infrecuente de predominio en la infancia causada por una respuesta inmunitaria anómala tras una infección faríngea por estreptococos beta-hemolíticos del grupo A. Dado el uso apropiado de antibióticos, su incidencia ha disminuido drásticamente en los últimos 50 años en el mundo occidental.
Clínica: Se caracteriza por la presencia de fiebre, artritis o carditis tras una faringitis estreptocócica. En un pequeño porcentaje de pacientes puede observarse clínica cutánea caracterizada por eritema marginado y nódulos subcutáneos53. Estos últimos son nódulos asintomáticos, frecuentemente asociados a afectación cardiaca, que aparecen sobre prominencias óseas.
Histopatología: Se observan depósitos subcutáneos de fibrina acompañados de un infiltrado variable de linfocitos, neutrófilos o células plasmáticas frecuentemente rodeado de una empalizada difusa de histiocitos. Estos datos histológicos no son patognomónicos de la enfermedad por lo que requieren una adecuada correlación clínico-patológica. El principal diagnóstico diferencial es el nódulo reumatoide, donde se aprecia una empalizada de histiocitos bien definida y un área central con densa necrobiosis (a diferencia de los nódulos subcutáneos reumáticos).
Nódulo reumatoideDefinición: Los nódulos reumatoides son lesiones inflamatorias cutáneas o subcutáneas que clásicamente se asocian a artritis reumatoide (AR).
Clínica: Aparecen hasta en un 30% de los pacientes con AR, constituyendo la manifestación extraarticular más frecuente de la enfermedad54,55. La localización más frecuente son zonas de presión o microtraumatismos y cerca de articulaciones. Las manos, los pies, las superficies extensoras de los brazos y las piernas (codos y rodillas), el cuero cabelludo, las orejas, los glúteos y la espalda son las más típicas54,55.
Se presentan como nódulos firmes, sin signos inflamatorios, que pueden estar adheridos a periostio y planos profundos. Su tamaño oscila entre 1 y 5cm. Generalmente son asintomáticos y no ulcerados salvo si se traumatizan. Su número es muy variable, aunque con más frecuencia son únicos o escasos (fig. 20A).
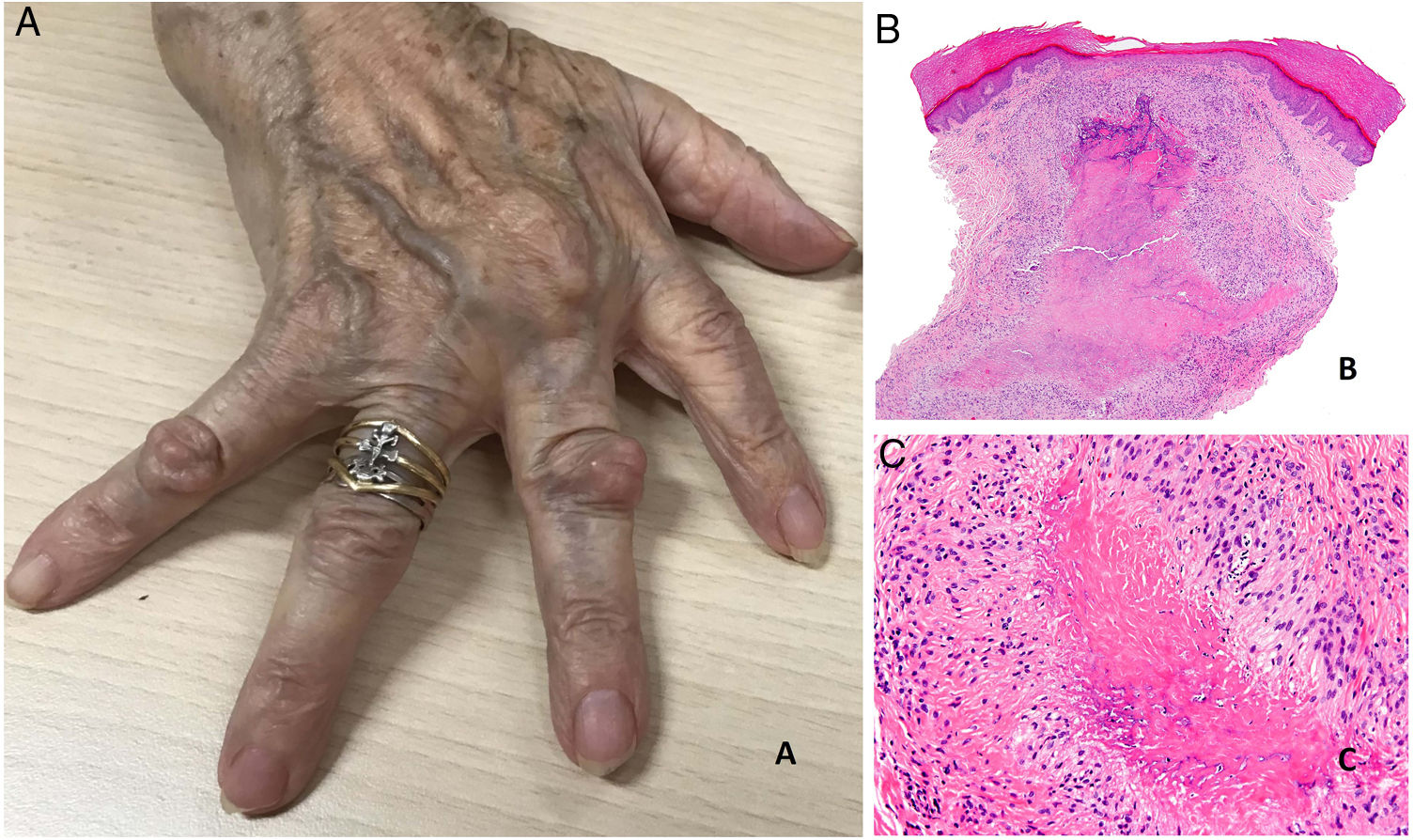
A) Artritis reumatoide. Aspecto clínico de 2 nódulos reumatoides en proximidad a las articulaciones interfalángicas de la mano. Tienen una superficie lisa y blanquecina, sin signos inflamatorios. B) Artritis reumatoide. Panorámica histológica de un nódulo reumatoide. En este caso la lesión alcanza zonas superficiales de la dermis y se extiende a tejidos profundos. Se reconoce una estructura de granuloma en empalizada con centro necrótico intensamente eosinófilo (H&E ×20). C) Artritis reumatoide. Detalle histológico del granuloma en empalizada que constituye la base histológica del nódulo reumatoide. Alrededor de un centro de necrosis fibrinoide de coloración intensamente eosinófila se dispone una capa de histiocitos en empalizada (H&E ×200).
Suelen asociarse a enfermedad articular severa, con títulos de factor reumatoide altos54,55. Sin embargo, se han descrito algunos pacientes con nódulos digitales múltiples que presentan una forma clínica más benigna, con escasa afectación articular, denominada «nodulosis reumatoide». El tratamiento con metotrexato puede inducir la aparición de nódulos múltiples lo que se ha denominado nodulosis reumatoide acelerada54,56.
Al igual que otras manifestaciones extraarticulares de la AR, los nódulos reumatoides no aparecen solamente en casos de enfermedad avanzada, sino que pueden presentarse en cualquier momento de la evolución, incluso en estadios tempranos55,56.
En raras ocasiones pueden aparecer nódulos reumatoides viscerales, especialmente en pulmones56.
Histopatología: El aspecto histológico es muy característico y representa una reacción granulomatosa de base inmunológica. Están constituidos por una zona central de necrosis, intensamente eosinófila debido al depósito de fibrina. A su alrededor se dispone un capa bien definida de histiocitos en empalizada rodeada con frecuencia de tejido de granulación54. Debido a su tendencia expansiva pueden tener aspecto multinodular confluente (figs. 20B y C). Su mayor tamaño, situación más profunda y coloración rojiza del centro necrótico, los distingue fácilmente del granuloma anular y otros tipos de granulomas cutáneos en empalizada. Pueden aparecer otros datos histológicos como fibrosis reactiva alrededor, calcificaciones distróficas y depósito de mucinas57. El perfil de citoquinas del nódulo reumatoide es muy similar al de la membrana sinovial de las articulaciones afectas por artritis reumatoide, lo que apoya una base autoinmune, aunque su patogenia permanece oscura56.
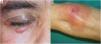
Acné agminataDefinición: También conocido como lupus miliar diseminado de la cara, el acné agminata es una dermatosis inflamatoria granulomatosa cuyo origen ha sido debatido durante años. Clásicamente se ha vinculado con entidades como la rosácea, la tuberculosis o la sarcoidosis58.
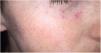
Clínica: Generalmente se presenta en pacientes jóvenes en forma de pápulas monomorfas eritematosas, marronáceas o amarillentas, localizadas en la zona central y laterales de la cara (fig. 21). La afectación de los párpados, especialmente de los inferiores, es muy habitual. En raras ocasiones puede aparecer en las axilas o en los miembros superiores. Suele ser un proceso autorresolutivo con cicatrices residuales secundarias, y responde irregularmente a tetraciclinas, isotretinoina, dapsona, clofazimina o isoniazida59.
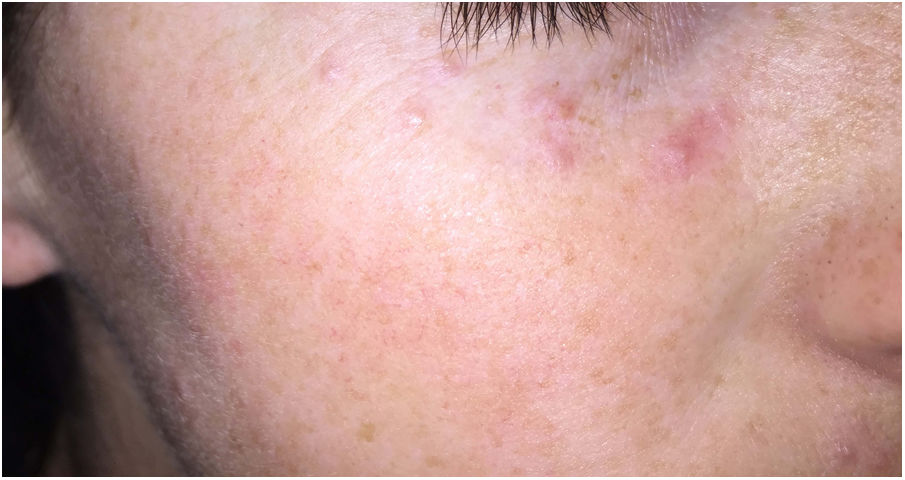
Histopatología: Se observa una respuesta granulomatosa epitelioide necrosante con necrosis caseosa central y evidencia de células gigantes multinucleadas (fig. 22). En ocasiones es posible ver esa necrosis en relación con un folículo piloso destruido. Con tinciones específicas se puede observar un anillo de fibras elásticas en el centro de un foco necrótico, lo que podría corresponderse con el istmo de un folículo piloso. No se ha establecido una relación con el Demodex folliculorum, como ocurre habitualmente en la rosácea58. La presencia de vasculitis focal es anecdótica.
Rosácea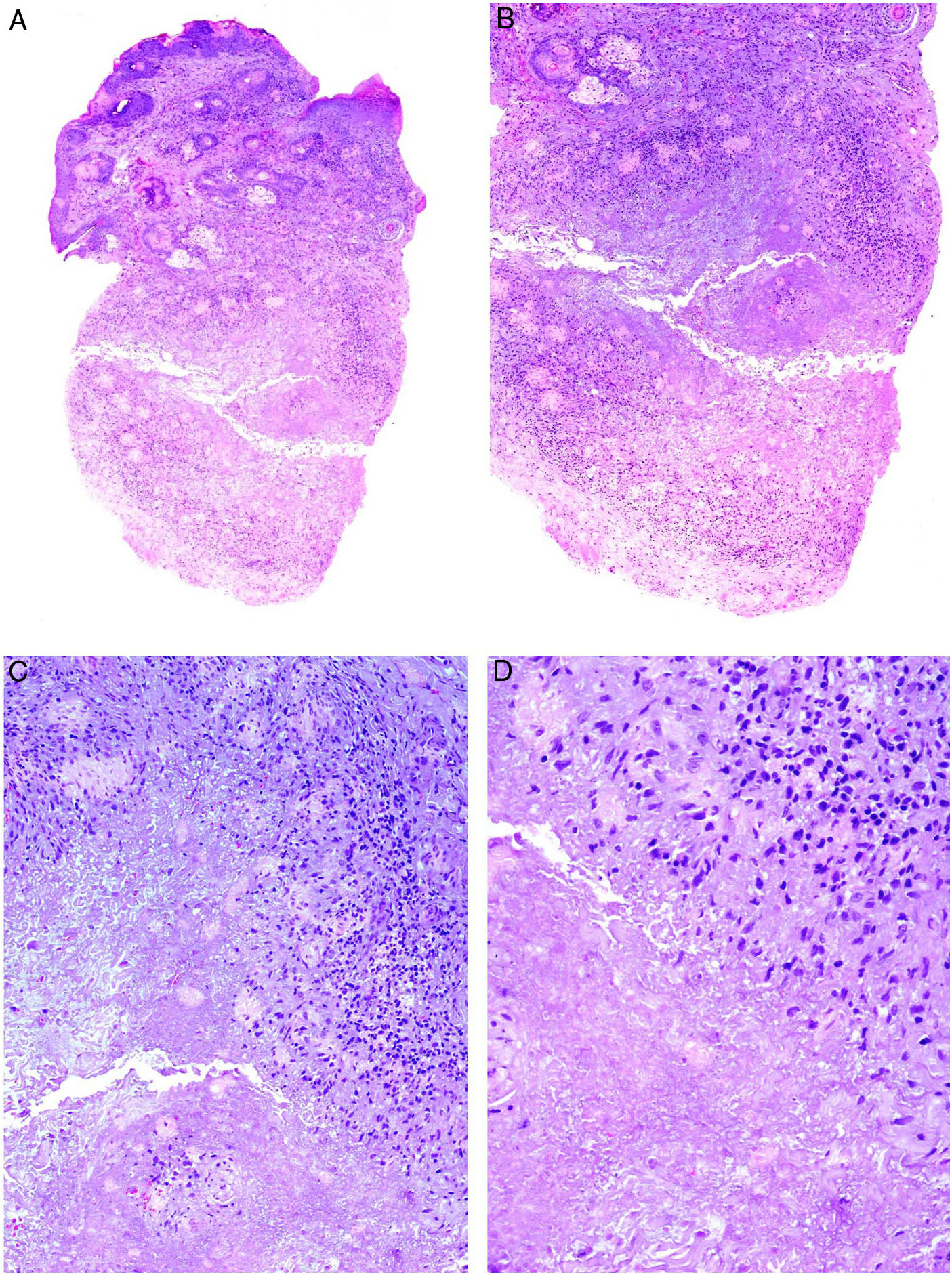
Definición: La rosácea es una enfermedad inflamatoria crónica de la unidad pilosebácea, que suele desarrollarse en la cara de pacientes de edad media y avanzada, en forma de pápulo-pustulosas acneiformes, con aumento de la reactividad vascular capilar al calor, lo que produce en la piel un eritema persistente y finalmente telangiectasias. En cuadros más avanzados, y sobre todo en varones, se puede acompañar, de hiperplasia sebacea, más llamativa en la nariz, lo que constituye el rinofima. Aunque la etiología es desconocida, el trastorno base parece ser una disminución del tono vascular con tendencia a la vasodilatación exagerada frente a determinados estímulos (cambios bruscos de frío a calor, el sol y los rayos UVA, bebidas alcohólicas, comidas muy calientes o picantes y factores estresantes), junto a la activación del sistema inmune e infestación por Demodex60,61.
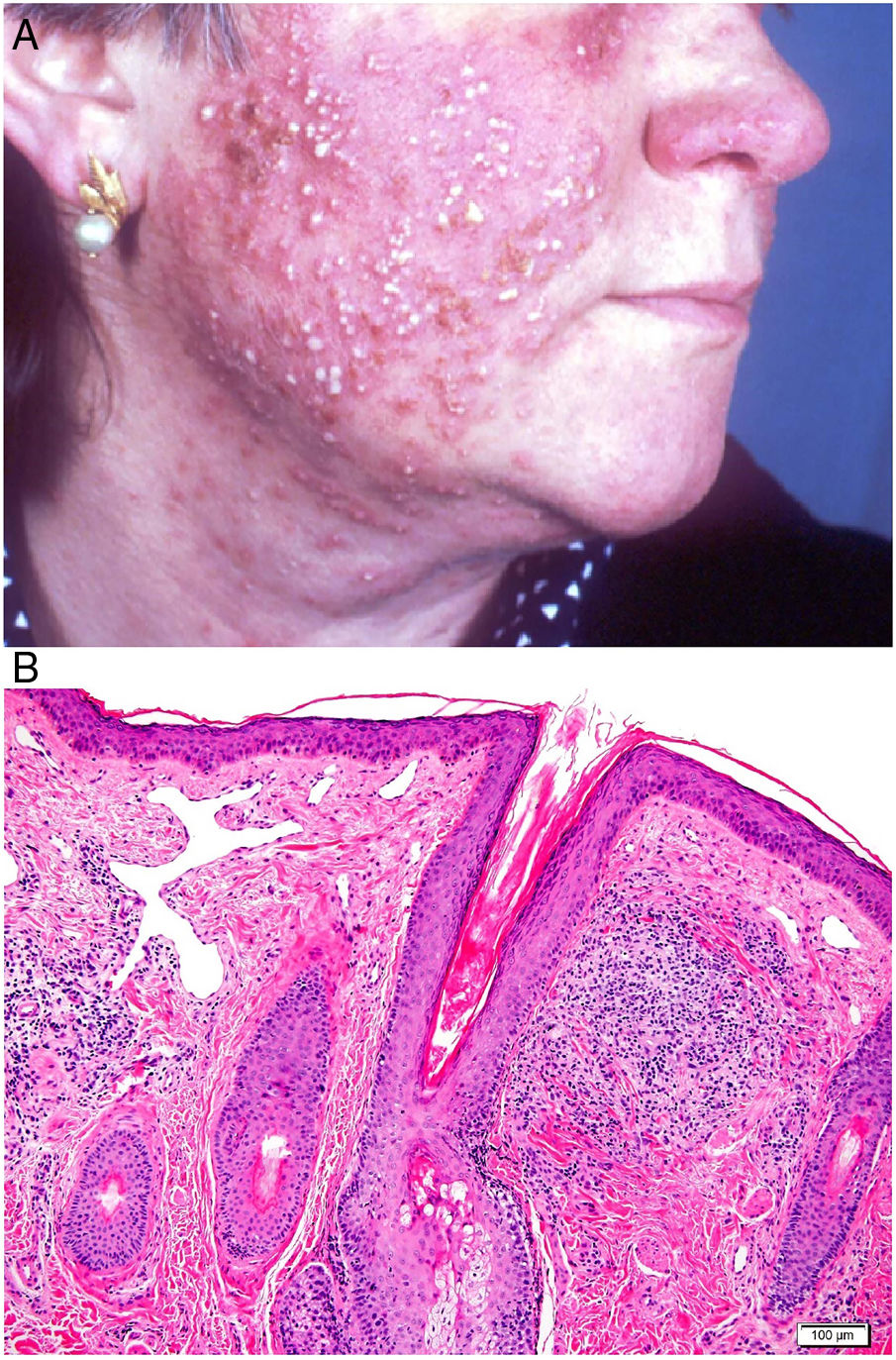
Clínica: Suele cursar en brotes agudos inflamatorios con tumefacción cutánea, cuya remisión espontánea es muy poco frecuente. Se describen 4 formas clínicas: telangiectásica, pápulopustulosa, granulomatosa y fimomatosa62,63. Los primeros signos clínicos son enrojecimiento y dilatación vascular en la nariz, mejillas y frente, inicialmente intermitente, aunque acaba por ser estable cuando surgen las telangiectasias. En su grado más avanzado aparecen pápulas y pústulas simétricas, característicamente sin comedones, aunque con prurito, escozor y ardor (fig. 23A). En ocasiones, además de las lesiones faciales, se pueden afectar los ojos, apareciendo blefaritis, conjuntivitis, epiescleritis, iritis y queratitis64,65. La forma granulomatosa cursa con pequeñas pápulas parduscas, induradas. El rinofima se caracteriza por masas lobuladas en punta y ala nasal en varones de más de 40 años, sobre las que se identifican marcada dilatación de los folículos pilosebáceos que aparecen ocluidos por grandes tapones de sebo y queratina.
El diagnóstico diferencial clínico, se debe por lo tanto realizar con enfermedades cutáneas que cursan con eritema facial como la dermatitis seborreica, el lupus eritematoso, la dermatomiositis, el síndrome carcinoide, la policitemia vera y las enfermedades cutáneas acneiformes como el acné vulgar, la dermatitis perioral, la foliculitis por Gram negativos y el acné corticoideo.
Histopatología: En las formas telangiectásicas solo se reconoce dilatación de los vasos linfáticos dérmicos, que se acompañan de infiltrado linfocitario y de células plasmáticas perivascular superficial. En las formas pápulo-pustulosas, el infiltrado es más denso y profundo, con presencia de abundantes neutrófilos, localizados alrededor de las estructuras vasculares y pilosebáceas. Se puede observar foliculitis aguda (en las lesiones pustulosas activas), tapones foliculares y Demodex (entre el 20 y el 50% de los casos). En la variante granulomatosa se advierten granulomas parafoliculares, constituidos por células epiteliodes con corona linfocitaria (fig. 23B), que pueden presentar necrosis central hasta en un 11% de los casos. El rinofima se caracteriza, además, por la presencia de hipertrofia de las glándulas sebáceas66.
El diagnóstico diferencial histopatológico incluye enfermedades granulomatosas oro-faciales entre las que destacan: la dermatitis perioral, la queilitis granulomatosa, la EC, la sarcoidosis, el lupus vulgar y la enfermedad granulomatosa crónica66. Las 2 primeras enfermedades son indistinguibles microscópicamente de la rosácea y, de hecho, algunos autores consideran que son formas relacionadas. Los granulomas de la EC son más profundos y afectan a la pared de los vasos. Los granulomas de la sarcoidosis son desnudos, sin corona linfocitaria. La presencia de bacilos ácido-alcohol resistentes es la clave diagnóstica del lupus vulgar. Por último, la enfermedad granulomatosa crónica se presenta en un contexto clínico de inmunodeficiencia en la población pediátrica.
Queilitis granulomatosa y síndrome de Melkersson-RosenthalDefinición: La queilitis granulomatosa (QG) es una enfermedad granulomatosa no necrosante infrecuente de los labios, mediada inmunológicamente. Se considera una forma monosintomática o incompleta del síndrome de Melkersson-Rosenthal (SMR).
El SMR es una rara enfermedad, caracterizada por la tríada de edema orofacial recidivante, parálisis facial y lengua fisurada.
Se emplea el término granulomatosis orofacial idiopática (GOF) para englobar las 2 anteriores, las cuales presentan unas características histológicas idénticas.
La sarcoidosis y sobre todo la EC pueden presentar características clínicas e histológicas semejantes a la GOF; sin embargo, una definición estricta de GOF debe descartar otras enfermedades granulomatosas sistémicas67.
Clínica: La presentación más habitual de la QG es la tumefacción aguda y asintomática de los labios, de carácter recurrente (fig. 24A). Con el tiempo la tumefacción se hace firme e indurada. Se pueden afectar otras zonas de la cara. El SMR, con frecuencia ocurre de forma incompleta, especialmente en el momento de la presentación. El diagnóstico puede requerir un largo seguimiento. Se ha sugerido el edema orofacial como síntoma indispensable para realizar el diagnóstico68.
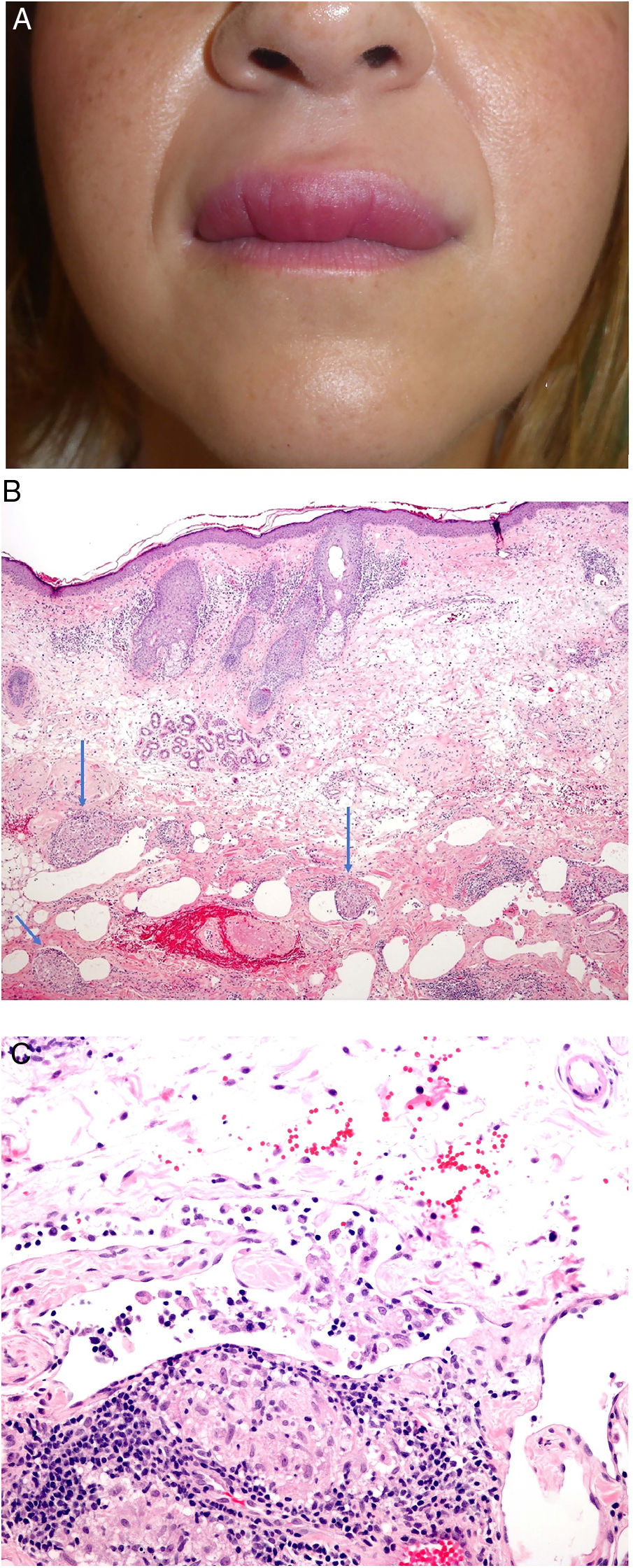
A) Queilitis granulomatosa. Marcado edema del labio superior. B) Queilitis granulomatosa. Piel de labio con marcado edema dérmico, ectasia vascular, discreta infiltración linfocitaria y granulomas epitelioides peri e intravasculares (flechas) (H&E ×40). C) Queilitis granulomatosa. Vaso linfático dilatado con agregados histiocitarios intravasculares y un granuloma perivascular (H&E ×200).
Histopatología: El epitelio de la mucosa labial o la epidermis muestran cambios reactivos con hiperqueratosis, acantosis y espongiosis con exocitosis linfocitaria. El corion o la dermis muestran edema con ectasia de vasos sanguíneos y linfáticos e infiltrados perivasculares e intersticiales linfocitarios o linfoplasmocitarios e histiocitarios. Más rara vez se reconocen eosinófilos. El hallazgo más característico (aunque no siempre presente) es la presencia de granulomas epitelioides no necrosantes; su frecuencia oscila entre el 43 y el 82% de los casos. Por otra parte, los granulomas a veces se localizan en la parte profunda de la biopsia y por tanto biopsias superficiales pueden no demostrar la inflamación granulomatosa.
Los granulomas suelen ser laxos y no compactos debido al edema intercelular entre las células epitelioides. Suelen ser pequeños, escasos en número, dispersos, no confluentes y sin anillo linfocitario69,70. Es frecuente que se dispongan adyacentes a vasos sanguíneos y linfáticos (fig. 24B). Más rara vez, se observan agregados histiocitarios o granulomas en el interior de vasos linfáticos dilatados (fig. 24C). Esta inflamación granulomatosa perivascular o intravascular es posiblemente la causa de la obstrucción linfática que conlleva al estasis linfático, la dilatación vascular y posteriormente al edema71,72. A veces, se identifican pequeños granulomas sarcoideos. En conclusión, es necesario realizar una correlación clínico-patológica para realizar este diagnóstico.
AutoríasTodos los autores firmantes han contribuido por igual en la elaboración del manuscrito, independientemente del orden de apellidos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este artículo es una iniciativa del Grupo Español de Dermatopatología de la Academia Española de Dermatología y Venereología y de la Sociedad Española de Anatomía Patológica.


