


La esclerosis tuberosa (ET) es un síndrome neurocutáneo infrecuente caracterizado por la aparición de hamartomas en múltiples órganos. Su diagnóstico se basa en criterios clínicos.
ObjetivoDescribir los hallazgos clínicos en una serie de 67 pacientes afectos de ET.
Material y métodosLlevamos a cabo un estudio retrospectivo, descriptivo y observacional de los pacientes con ET remitidos a nuestras consultas de Dermatología entre enero de 1994 y marzo de 2007.
ResultadosEl 100 % de los pacientes presentaron alteraciones neurológicas o dermatológicas. El resto fueron, por orden: psiquiátricas (55,5 %), renales (32,8 %), cardíacas (22,4 %), esqueléticas y pulmonares (13,4 %) y oftalmológicas (11,9 %).
ConclusionesDescribimos los hallazgos clínicos en una serie de pacientes afectos de ET. Se trata, según la literature revisada, del primer estudio de este tipo en la población española. Globalmente, nuestros datos apoyan lo hasta ahora publicado.
Tuberous sclerosis is an uncommon neurocutaneous syndrome characterized by the appearance of hamartomas in multiple organs. Diagnosis is based on clinical criteria.
ObjectiveTo report the clinical findings in a series of 67 patients with tuberous sclerosis.
Material and methodsThis was a descriptive and observational retrospective study of patients with tuberous sclerosis referred to our dermatology clinics between January 1994 and March 2007.
ResultsAll patients presented neurological or dermatological disorders. Other disorders, in descending frequency, were psychiatric (55.5 %), renal (32.8 %), cardiac (22.4 %), skeletal and pulmonary (13.4 %), and ophthalmological (11.9 %).
ConclusionsWe report the clinical findings in a series of patients with tuberous sclerosis. According to our literature search, this is the first such study in the Spanish population. Overall, our findings support those already published.
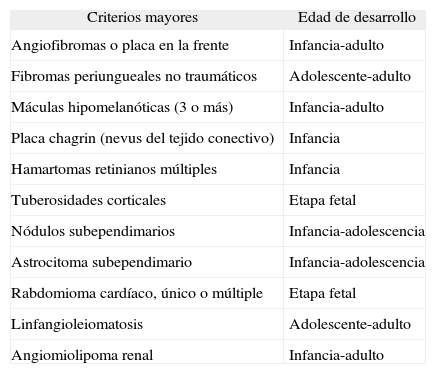
La esclerosis tuberosa (ET) es un síndrome neurocutáneo de herencia autosómica dominante, caracterizado por la aparición de hamartomas en múltiples órganos. La tríada clínica clásica engloba angiofibromas (AF), retraso mental y epilepsia; sin embargo, esta sólo aparece en el 29 % de los pacientes y el 6 % no tiene ninguno de los signos1. La incidencia de ET se estima entre 1/5.800 y 1/10.000 y en dos tercios de los pacientes se debe a mutaciones esporádicas2. Se han identificado dos genes responsables de la enfermedad: TSC1 en el cromosoma 9 y TCS2 en el cromosoma 163. La enorme variabilidad clínica de la ET se explica por mutaciones en estos genes y mosaicismos genéticos de ambos, así como una penetrancia variable. El diagnóstico de la ET es fundamentalmente clínico y se basa en una serie de criterios (tabla 1), ya que no existe un único hallazgo diagnóstico4. La principal causa de morbimortalidad de la ET se debe a las manifestaciones neurológicas2.
Criterios diagnósticos de esclerosis tuberosa4
| Criterios mayores | Edad de desarrollo |
| Angiofibromas o placa en la frente | Infancia-adulto |
| Fibromas periungueales no traumáticos | Adolescente-adulto |
| Máculas hipomelanóticas (3 o más) | Infancia-adulto |
| Placa chagrin (nevus del tejido conectivo) | Infancia |
| Hamartomas retinianos múltiples | Infancia |
| Tuberosidades corticales | Etapa fetal |
| Nódulos subependimarios | Infancia-adolescencia |
| Astrocitoma subependimario | Infancia-adolescencia |
| Rabdomioma cardíaco, único o múltiple | Etapa fetal |
| Linfangioleiomatosis | Adolescente-adulto |
| Angiomiolipoma renal | Infancia-adulto |
| Criterios menores |
| Depresiones dentales múltiples |
| Hamartoma rectal polipoideo |
| Quistes óseos |
| Alteraciones en la migración de la sustancia blanca |
| Fibromas gingivales |
| Hamartomas no renales |
| Manchas acrómicas en la retina |
| Hipopigmentación en confeti |
| Quistes renales múltiples |
Diagnóstico definitivo: dos criterios mayores o uno mayor y dos menores; probable: uno mayor y otro menor; posible: uno mayor y dos o más menores.
En este trabajo describimos los hallazgos en una serie de 67 pacientes y comparamos nuestros resultados con los de la literatura revisada.
Material y métodosLlevamos a cabo un estudio retrospectivo, descriptivo y observacional de los pacientes con ET remitidos a nuestras consultas de Dermatología entre enero de 1994 y marzo de 2007. Se incluyeron los pacientes que cumplían, según los criterios diagnósticos, un diagnóstico definitivo de ET (tabla 1). Los datos de los pacientes se extrajeron de una base de datos Excel® y se complementaron con la historia clínica y los estudios oftalmológicos, neurológicos, psiquiátricos, etc., a los que fueron sometidos. Se describe la edad, el sexo y los antecedentes familiares de ET de los pacientes. En cuanto a las alteraciones asociadas se recogieron en todos ellos, clasificándolas en diferentes grupos que incluyen: alteraciones dermatológicas, patología cardíaca, patología del sistema nervioso central (SNC), trastornos psiquiátricos, alteraciones renales, óseas y pulmonares y trastornos oftalmológicos.
Las manifestaciones dermatológicas se estudiaron en mayor detalle:
- 1.
Los AF se clasificaron según su localización en la cara (mejillas, nariz, surco nasogeniano y frente), su patrón de presentación (en empedrado o sebáceo) y distribución (unilateral o bilateral) (fig. 1). Se define el patrón sebáceo como la aparición de lesiones aisladas, separadas unas de otras, y el patrón en empedrado cuando confluyen formando una placa con múltiples lobulaciones en su superficie.
- 2.
Las manchas hipomelanóticas se clasificaron en 4 grupos: lanceolada o en hoja de fresno, en confeti, dactilar y en otras (ninguno de los tres grupos anteriores). Asimismo se consideró su localización en tórax anterior, en tórax posterior, en miembro superior, miembro inferior y abdomen.
- 3.
En los pacientes que presentaron tumores de Koenen se tuvo en cuenta si aparecían en las manos o en los pies. Las placas«chagrin»o«piel de zapa»se clasificaron también según su localización.

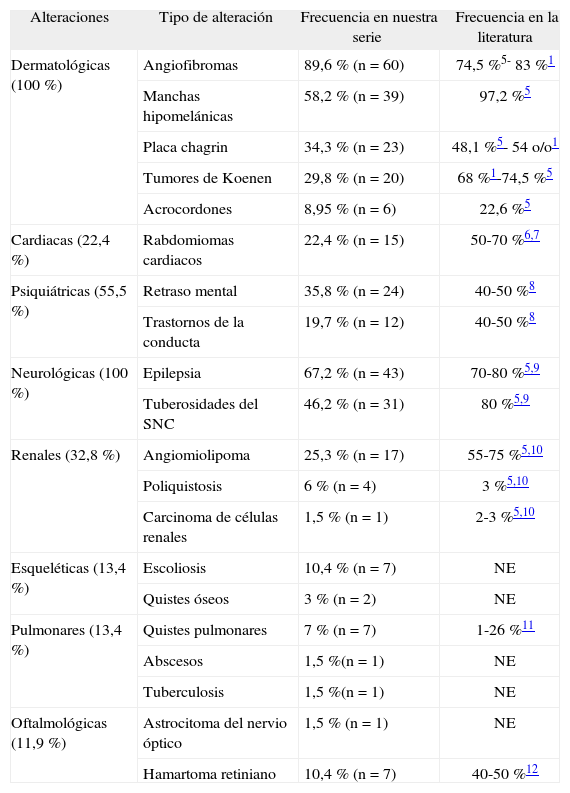
Se recopilaron 67 pacientes, 35 mujeres y 32 varones, con una edad media de 27,61 años (rango de 15 a 80 años). El 21 % tenía antecedentes familiares de ET. Los resultados se resumen en la tabla 2. El 100 % de los mismos presentaba alteraciones dermatológicas; de ellas las más frecuentes fueron los AF en el 89,6 %, seguidos de las manchas hipomelanóticas en un 58,2 %, las placas chagrin en un 34,3 %, los tumores de Koenen en un 29,8 % y los acrocordones en un 8,95 %. De los 67 pacientes 15 presentaron alteraciones cardíacas, todas ellas en forma de rabdomioma (22,4 %). Las alteraciones psiquiátricas aparecieron en un 55,5 % de los pacientes, en forma de retraso mental en un 35,8 % y trastornos de la conducta en un 19,7 %. Un 100 % de los pacientes presentó alteraciones neurológicas, el 67,2 % se diagnosticó de epilepsia y el 46,2 % presentaba tuberosidades en el SNC en las pruebas de imagen. Las alteraciones renales se encontraron en un 32,8 %, un 25,3 % presentaba angiomiolipomas, un 6 % poliquistosis renal y en un paciente (1,5 %) se diagnosticó un carcinoma de células renales. Un total de 9 sujetos presentaron alteraciones esqueléticas, en 7 escoliosis (10,4 %) y en 2 quistes óseos (3 %). Se apreciaron alteraciones pulmonares en 9 pacientes, en 7 se objetivaron quistes simples (10,4 %), uno tuvo un absceso (1,5 %) y otro tuberculosis (1,5 %). Las alteraciones oftalmológicas se diagnosticaron en 10 pacientes, 7 con hamartomas retinianos (10,4 %) y tres con astrocitomas del nervio óptico (4,47 %).
Alteraciones encontradas en los pacientes
| Alteraciones | Tipo de alteración | Frecuencia en nuestra serie | Frecuencia en la literatura |
| Dermatológicas (100 %) | Angiofibromas | 89,6 % (n=60) | 74,5 %5- 83 %1 |
| Manchas hipomelánicas | 58,2 % (n=39) | 97,2 %5 | |
| Placa chagrin | 34,3 % (n=23) | 48,1 %5- 54 o/o1 | |
| Tumores de Koenen | 29,8 % (n=20) | 68 %1-74,5 %5 | |
| Acrocordones | 8,95 % (n=6) | 22,6 %5 | |
| Cardiacas (22,4 %) | Rabdomiomas cardiacos | 22,4 % (n=15) | 50-70 %6,7 |
| Psiquiátricas (55,5 %) | Retraso mental | 35,8 % (n=24) | 40-50 %8 |
| Trastornos de la conducta | 19,7 % (n=12) | 40-50 %8 | |
| Neurológicas (100 %) | Epilepsia | 67,2 % (n=43) | 70-80 %5,9 |
| Tuberosidades del SNC | 46,2 % (n=31) | 80 %5,9 | |
| Renales (32,8 %) | Angiomiolipoma | 25,3 % (n=17) | 55-75 %5,10 |
| Poliquistosis | 6 % (n=4) | 3 %5,10 | |
| Carcinoma de células renales | 1,5 % (n=1) | 2-3 %5,10 | |
| Esqueléticas (13,4 %) | Escoliosis | 10,4 % (n=7) | NE |
| Quistes óseos | 3 % (n=2) | NE | |
| Pulmonares (13,4 %) | Quistes pulmonares | 7 % (n=7) | 1-26 %11 |
| Abscesos | 1,5 %(n=1) | NE | |
| Tuberculosis | 1,5 %(n=1) | NE | |
| Oftalmológicas (11,9 %) | Astrocitoma del nervio óptico | 1,5 % (n=1) | NE |
| Hamartoma retiniano | 10,4 % (n=7) | 40-50 %12 |
NE: no especificada; SNC: sistema nervioso central.
El estudio de las alteraciones dermatológicas se resume en la tabla 3. Los AF aparecieron en 60 pacientes y la localización más frecuente fue en las mejillas (78,3 %). En menor proporción se localizaron en la nariz (48,3 %), en el mentón (33,3 %) y en el surco nasogeniano y la frente (13,3 %). El patrón más frecuente de presentación fue en empedrado en el 80 % y el 20 % de los AF fue de patrón sebáceo. La gran mayoría se distribuía de forma bilateral (98,33 %), aunque en uno de los pacientes lo hacía de manera unilateral. Las manchas hipomelanóticas aparecieron en 39 de los 67 pacientes. La forma más frecuente fue la lanceolada o en hoja de fresno (76,92 %), seguidas de las manchas en confeti (51,28 %) y dactilares (30,76 %). Un 40 % de las manchas hipomelanóticas no se pudieron clasificar en ninguno de los grupos anteriores. La localización más habitual fue en el tórax anterior (58,97 %), en los miembros inferiores (56,41 %), en los superiores (33,33 %), en el tórax posterior (12,82 %) y en el abdomen (10,25 %). Los tumores de Koenen se apreciaron en 20 pacientes, con afectación similar de las manos (60 %) y los pies (55 %). La placa en piel de zapa o placa chagrin se apreció en 23 pacientes, en la mayoría de localización lumbar (73,91 %), aunque también apareció en la región dorsal (21,73 %).
Descripción de las alteraciones dermatológicas
| Angiofibromas 89,6 % (n=60) | Localización | Mejillas: 78,3 % (n=47) |
| Nariz: 48,3 % (n=29) | ||
| Mentón: 33,3 % (n=20) | ||
| Surco nasogeniano, frente: 13,3 % (n=8) | ||
| Patrón de presentación | Empedrado: 80 % (n=48) | |
| Sebáceos: 20 % (n=12) | ||
| Distribución | Bilateral: 98,33 %(n=59) | |
| Unilateral: 1,6 % (n=1) | ||
| Manchas hipomelanóticas 58,2 % (n=39) | Forma | Lanceolada o en hoja de fresno: 76,92 % (n=30) |
| Confeti: 51,28 % (n=20) | ||
| Dactilar: 30,76 % (n=12) | ||
| Otras: 40 % (n=24) | ||
| Localización | Tórax anterior: 58,97 % (n=23) | |
| Miembros inferiores: 56,41 % (n=22) | ||
| Miembros superiores: 33,33 %(n=13) | ||
| Tórax posterior: 12,82 %(n=5) | ||
| Abdomen: 10,25 %(n=4) | ||
| Tumores de Koenen 29,8 % (n=20) | Localización | Manos: 60 % (n=12) |
| Pies: 55 %(n=11) | ||
| Placa Chagrin 34,3 % (n=23) | Localización | Lumbar: 73,91% (n=17) |
| Tronco posterior: 21,73 % (n=5) |
El término esclerosis tuberosa fue acuñado en 1880 por Bourneville 13. En 1908 Vogt describe la tríada clásica de retraso mental, crisis epilépticas y AF faciales14, presente sólo en un 29 % de los pacientes1. Desde entonces se han publicado varias series de pacientes evaluando la frecuencia de las manifestaciones clínicas de la enfermedad. De lo hasta ahora publicado se deduce un amplio espectro clínico en su presentación. En la última década el descubrimiento de los genes implicados (TSC1 y TSC2) y los modelos observados en la Drosophila melanogaster, la mejor descripción de los mosaicismos genéticos y la variable penetración de la enfermedad, ayudan a comprender por qué existen pacientes con mínimos signos y otros con una importante afectación.
El diagnóstico de ET se basa en una serie de criterios mayores y menores, ya que ningún criterio clínico aislado es diagnóstico. Las manifestaciones clínicas de la ET aparecen en distintos momentos del desarrollo (tabla 1)4; por tanto, la validez de estos criterios para el diagnóstico precoz está limitada por el hecho de que algunos tengan lugar en la infancia tardía o la adolescencia, sumado a los múltiples casos atípicos de presentación. Así, los rabdomiomas cardíacos y las tuberosidades corticales aparecen en la etapa fetal. Las lesiones cutáneas se aprecian en el 90 % de los pacientes de cualquier edad. Las primeras en detectarse son las máculas hipomelanóticas en la primera infancia, mientras que la placa chagrin suele aparecer después de los 5 años de edad. Los AF faciales pueden hacerlo a cualquier edad, pero fundamentalmente en la infancia tardía. Los fibromas periungueales tienden a surgir tras la pubertad. Los astrocitomas subependimarios se pueden desarrollar en la infancia y en la adolescencia, los angiomiolipomas renales en la infancia temprana o la adolescencia y la linfangioleiomatosis en las mujeres en la adolescencia tardía o en la edad adulta. La edad media de nuestra muestra es de 27,6 años, lo que lleva a suponer que han aparecido ya en los pacientes la gran mayoría de las manifestaciones cutáneas.
En nuestra serie el 100 % tiene alteraciones dermatológicas. Por tanto, el realizar un detallado examen dermatológico en los pacientes en los que se sospeche ET es altamente rentable, a la vez que accesible y fácil de llevar a cabo. El hallazgo más frecuente en nuestros pacientes fueron los AF faciales (89,6 %), mientras que en la literatura revisada son las manchas hipomelanóticas. Jozwiak5, en una serie de 106 niños con ET, encuentra manchas hipomelanóticas en un 97,2 %. En nuestro grupo de pacientes aparecen en el 58,2 %. La placa de chagrin la encontramos en el 34,3 %, con frecuencias similares a las de la literatura (48-54 %)1,5. Sin embargo, los tumores de Koenen y los acrocordones se observan en frecuencias inferiores a lo publicado (29,8 y 8,95 %, respectivamente).
El estudio en detalle de las lesiones dermatológicas no aparece en la literatura revisada. La forma más frecuente de presentación de los AF en nuestra muestra fue en las mejillas (78,3 %), con patrón en empedrado (80 %) y de forma bilateral (98,33 %). Otras localizaciones menos frecuentes fueron la nariz (48,3 %) y el mentón (33,3 %). Existe un caso de presentación de las lesiones de forma unilateral, probablemente como manifestación de un mosaicismo segmentario tipo I15, ya que todos los pacientes fueron diagnosticados formalmente de ET. Estas lesiones cutáneas provocan al paciente importantes problemas estéticos, psicológicos o médicos. En la actualidad el tratamiento de primera elección de los AF es el láser, ya que permite la destrucción más selectiva de las lesiones, minimizando el daño térmico residual y, por tanto, los efectos secundarios. Para aquellos AF en los que predomina el componente fibroso o para los AF protuberantes, el láser de CO2 en sus diferentes modalidades constituye el tratamiento de elección. Recientemente hemos publicado un estudio retrospectivo realizado en nuestro centro16, en el que valoramos los resultados a largo plazo del tratamiento con láser de CO2 (continuo y superpulsado) de los AF. En él se pone de manifiesto el alto porcentaje de recidivas (60,9 %), a pesar de las buenas y excelentes respuestas iniciales. Las recidivas aparecieron más precozmente cuando los pacientes eran tratados antes de los 20 años.
Las manchas hipomelanóticas halladas con más frecuencia fueron en forma de hoja de fresno (76,92 %), como se describen clásicamente en la enfermedad. Menos habituales fueron las formas en confeti (51,28 %) o dactilares (30,76 %). Existe un 40 % de manchas no clasificable en las formas anteriores, indicando nuevamente la variabilidad clínica de esta enfermedad. La localización más común fue en el tórax anterior y en los miembros. Los tumores de Koenen se hallaron de forma más o menos uniforme en las manos y los pies (60 y 55 % respectivamente). La localización de la placa Chagrin es más frecuente, tal y como describen los textos clásicos, en la región lumbar (73,91 %); sin embargo, algunas se localizan en zonas más superiores como el tronco posterior (21,3 %).
Las alteraciones neurológicas, al igual que las dermatológicas, aparecen en el 100 % de los pacientes de nuestra serie. Son la principal causa de morbimortalidad en los pacientes afectos de ET. Incluyen epilepsia y el hallazgo de tuberosidades corticales por técnicas de neuroimagen (tomografía axial computarizada [TAC] o resonancia magnética nuclear [RMN]), ambas íntimantente relacionadas, al ser las tuberosidades la causa de las crisis. Las tuberosidades son alteraciones del desarrollo del córtex cerebral, en las que histológicamente se pierde la distribución normal en 6 capas y aparecen neuronas dismórficas, astrocitos grandes y un tipo especial de célula conocido como célula gigante17. Persisten a lo largo de la vida y su comportamiento es benigno, salvo la sintomatología que puedan ocasionar. Según la literatura, hasta el 70-80 % de los pacientes con ET presenta epilepsia5,9 y el 80 %5,9 tuberosidades corticales. En nuestra serie de pacientes los porcentajes de nuevo son inferiores, con un 67,2 % con diagnóstico de epilepsia y un 46,2 % con hallazgos de tuberosidades en el SNC.
Las alteraciones psiquiátricas son las terceras más prevalentes en nuestra muestra de pacientes y se detectan hasta en un 55,5 % de los mismos. El 35,8 % de los sujetos está diagnosticado de retraso mental y casi el 20 % de trastornos de la conducta. En la literatura estos hallazgos se sitúan en torno al 40-50 %8; aunque este porcentaje se refiere globalmente a aquellos con alteraciones psicológicas y del desarrollo intelectual y no está desglosado como el de nuestro grupo. Este tipo de trastornos también se relaciona estrechamente con la aparición de tuberosidades corticales, especialmente de localización frontal. El trastorno del comportamiento más frecuentemente asociado a la ET es el autismo5.
Determinadas alteraciones renales están estrechamente asociadas con la ET. El angiomiolipoma es la manifestación más frecuente, ya que aparece en un 55-75 % de los pacientes. Los angiomiolipomas son tumores benignos compuestos por vasos aberrantes, células de músculo liso inmaduras y células grasas. En algunos pacientes con ET son bilaterales y múltiples10. Se detectan por ecografía, TAC o RMN. Su principal complicación, sobre todo en los mayores de 3cm, es el sangrado5,10. En nuestro grupo de pacientes se detecta en el 25,3 %. Otros hallazgos son poliquistosis renal en un 6 % (3 % en la literatura) y carcinoma de células renales en un 1,5 % (2-3 % en la literatura). La incidencia de carcinoma de células renales en la población general es similar a la de los pacientes con ET, si bien en estos se suele diagnosticar a edades más tempranas10.
El rabdomioma cardiaco es una tumoración intracavitaria o intramural que se ha detectado hasta en el 50-70 % de los pacientes, pero causa problemas en un porcentaje bastante inferior de los mismos. Se suele presentar en forma de insuficiencia cardiaca en la infancia y taquiarritmias. Es una de las primeras manifestaciones de la ET, de hecho se puede detectar intraútero, siendo uno de los principales marcadores de diagnóstico prenatal de ET. Es, además, el tumor cardiaco que se detecta con mayor frecuencia en el periodo neonatal7. En nuestros pacientes aparece en un porcentaje inferior, del 22,4 %.
La lesión pulmonar más comúnmente asociada es la linfangioleiomatosis, que aparece en mujeres afectas de ET en la segunda o tercera década de la vida. Se caracteriza por la proliferación de células musculares lisas que provocan espacios quísticos y neumotórax. Se ha observado empeoramiento durante el embarazo y la administración de estrógenos5,18. En la literatura aparece en un 57 % de los pacientes afectados de ET11, mientras que en nuestra serie se diagnostica sólo en el 7 %. En nuestro grupo sólo se estudia a los sintomáticos, por lo que no se identifica a aquellos que presentan cambios subclínicos. Este hecho puede justificar su hallazgo en porcentajes inferiores.
Los hamartomas en la retina son la alteración ocular que se asocia con más frecuencia a la ET y aparecen en un 40-50 % de los pacientes. En la mayoría permanecen estables y asintomáticos toda la vida12. En nuestra serie se da en un número inferior, el 10,4 %, quizá sea por la edad media de nuestros pacientes, 27 años, ya que, precisamente, la incidencia de hamartomas retinianos aumenta con la edad de los sujetos.
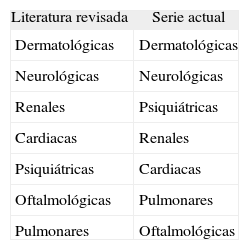
En general, en nuestra serie de pacientes los hallazgos clínicos son inferiores a lo que aparece en la literatura revisada. Puede ser debido a las propias limitaciones del estudio (estudio retrospectivo, los métodos de la recopilación de casos y el número limitado de pacientes). Por otro lado, la ET es una enfermedad clínicamente muy polimorfa con un amplio espectro de afectación, desde casos muy leves a graves, que además varían en su expresión a lo largo del tiempo, lo que podría explicar los diferentes porcentajes hallados. No obstante, nuestra serie constituye una investigación realizada en la práctica clínica y los pacientes, con determinadas afectaciones, no están uniformemente estudiados, ya que las pruebas complementarias se realizan de forma dirigida en función de los síntomas. Sin embargo, si ordenamos la frecuencia de las alteraciones asociadas en nuestra serie de pacientes y la literatura revisada (tabla 4) las diferencias son mínimas, corroborando al menos un orden de frecuencias.
Orden en frecuencia de las manifestaciones de la esclerosis tuberosa según la literatura y según nuestra serie de pacientes
| Literatura revisada | Serie actual |
| Dermatológicas | Dermatológicas |
| Neurológicas | Neurológicas |
| Renales | Psiquiátricas |
| Cardiacas | Renales |
| Psiquiátricas | Cardiacas |
| Oftalmológicas | Pulmonares |
| Pulmonares | Oftalmológicas |
Describimos los hallazgos en una serie de pacientes con ET y su comparación con las hasta ahora publicadas. No se han realizado muchos estudios de este tipo, siendo este el primero en la población española; sin embargo, a pesar de pequeñas diferencias apreciadas, globalmente nuestro trabajo apoya lo publicado hasta ahora.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.


