





INTRODUCCION
La elastosis perforante serpiginosa (EPS) es una dermatosis crónica e infrecuente, que pertenece al grupo de las dermatosis perforantes primarias. Probablemente los primeros casos fueron descritos en 1953 por Lutz1 con el nombre de queratosis folicular serpiginosa. En 1968, Mehregan2 publicó la primera revisión, delimitándola clínica e histopatológicamente.
Es más frecuente en varones, sobre todo entre la segunda y tercera décadas de la vida. Entre los casos idiopáticos se han publicado casos familiares, sugiriéndose una herencia autosómica dominante, con expresividad variable3,4. Se han descrito formas asociadas con el síndrome de Down (tabla 1)5,6 y con trastornos hereditarios del tejido conjuntivo (tabla 2), formas inducidas por el tratamiento crónico con penicilamina, y formas idiopáticas7. Todas ellas son similares desde el punto de vista clínico, con pápulas centradas por un tapón de queratina, que adoptan formas arciformes, y que en su evolución dejan una atrofia central e hipopigmentaciones residuales. Es frecuente el fenómeno de Koebner. Son lesiones asintomáticas, o levemente pruriginosas, que suelen localizarse en la cara, nuca, caras laterales del cuello y/o extremidades superiores, con una distribución típicamente simétrica. Se ha descrito de forma ocasional la resolución espontánea de las lesiones8.


Presentamos un caso de EPS en un paciente con síndrome de Down, con una localización poco común en las extremidades inferiores.
DESCRIPCION DEL CASO
Un varón de 22 años con síndrome de Down, sin antecedentes medicoquirúrgicos de interés, acudió a nuestra consulta por la aparición progresiva desde hacía 4 años, de unas lesiones muy pruriginosas suprapatelares bilaterales. La anamnesis detallada por aparatos no reveló otra sintomatología asociada. Ningún miembro de la familia había tenido lesiones similares previamente.
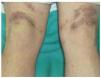
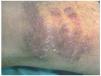
En la exploración física se observaron, en la cara anterior de ambos muslos, y en la cara posterior del muslo izquierdo (fig. 1), unas pápulas eritematovioláceas de 2-3 mm de diámetro, con una hiperqueratosis marcada en su región central. Algunas aparecían aisladas, pero la mayoría se localizaban en la periferia de unas placas de morfología serpiginosa, con un centro discretamente atrófico (fig. 2).
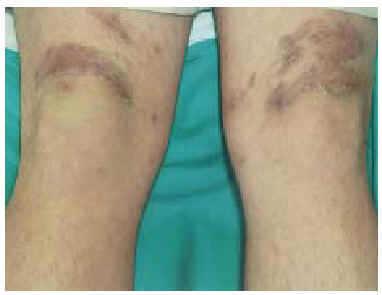
Fig. 1.--Distribución simétrica de las lesiones en las extremidades inferiores.
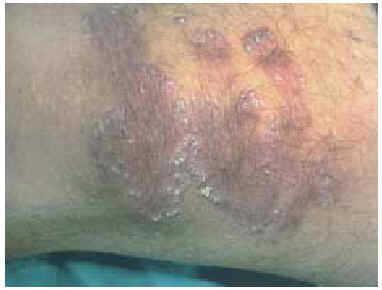
Fig. 2.--Pápulas hiperqueratósicas adoptando una morfología serpiginosa.
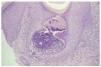
Realizamos una biopsia cutánea, en la que pudieron observarse conductos estrechos y sinuosos que comunicaban la dermis con la superficie, ocupados por restos celulares basófilos y fibras elásticas eosinófilas de grosor variable. Encontramos unos tapones de queratina ortoqueratósica en la salida de estos conductos, junto a una epidermis hiperplásica con acantosis irregular (fig. 3). En la dermis superficial existía un claro incremento en el número de las fibras elásticas, gruesas y eosinófilas, y un discreto infiltrado de histiocitos y células gigantes multinucleadas tipo cuerpo extraño. Al ser estas fibras autofluorescentes, pudieron visualizarse mejor con el microscopio de fluorescencia, de un color amarillo-verdoso brillante (fig. 4).

Fig. 3.--Epidermis hiperplásica, junto al canal epidérmico relleno de fibras elásticas eosinófilas y detritos celulares basófilos. (Hematoxilina-eosina, x200.)
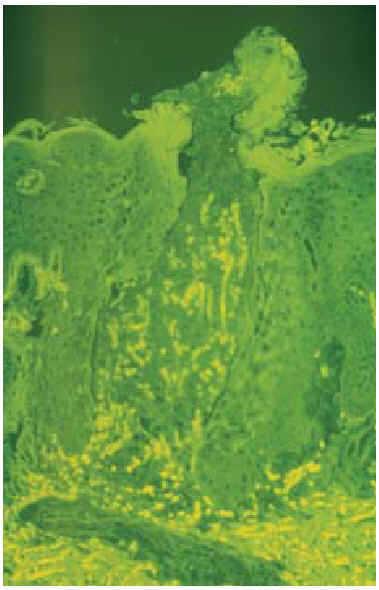
Fig. 4.--Fibras elásticas eliminándose a través del canal epidérmico. Microscopio de fluorescencia. (Hematoxilina-eosina, x200.)
Se comenzó el tratamiento con mometasona y calcipotriol tópicos, sin obtener mejoría tras 6 meses de seguimiento, durante los cuales siguieron apareciendo lesiones. A continuación se asoció vaselina salicílica al 10 % en aplicación tópica, dos veces al día, y acitretina por vía oral, 20 mg diarios, con buenos controles clínicos y analíticos. Con esta nueva pauta no han aparecido lesiones nuevas y se han atenuado las existentes, durante 7 meses de tratamiento.
DISCUSION
El desencadenante de la EPS es la producción de un exceso de fibras elásticas alteradas morfológica y bioquímicamente, que con frecuencia se asocia con alteraciones coexistentes de las fibras colágenas. Las fibras elásticas alteradas actúan como material extraño, y desencadenan una reacción por la que son eliminadas a través de conductos transepidérmicos9. Los individuos con síndrome de Down tienen frecuentes problemas inmunológicos, y se ha sugerido que en estos pacientes la alteración de la función fagocítica podría facilitar la eliminación epidérmica, como una alternativa para expulsar estos detritos celulares8,10. Jan et al11 describieron un posible agravamiento de las lesiones por déficit de vitamina A en un paciente con trisomía del cromosoma 21 y EPS.
Algunos autores han propuesto un mecanismo inmunológico. Las fibras elásticas anormales localizadas en la dermis papilar pueden representar un estímulo antigénico, que desencadenaría una respuesta inflamatoria, que a su vez estimularía la eliminación de esas fibras elásticas8. Apoya este hecho encontrar depósitos de inmunoglobulinas (IgG, IgA, IgM) y C3 en la dermis papilar12. Sin embargo, no hay estudios posteriores que confirmen estos hallazgos.
El mecanismo molecular de la eliminación transepidérmica de las fibras dérmicas alteradas está pobremente definido, aunque recientemente Fujimoto et al13,14 han demostrado la inducción de la expresión del receptor de elastina de 67 kDa en los queratinocitos que rodean la región de la eliminación de las fibras elásticas, lo que implica una interacción entre el queratinocito y la elastina en este proceso. Otros autores han propuesto que los valores elevados de fibronectina en el suero y en la piel, son los responsables de incitar un aumento de la migración y proliferación epitelial, facilitando la eliminación transepidérmica15.
En nuestro paciente destaca la localización poco frecuente de la EPS en las extremidades inferiores, reflejado escasamente en la literatura médica11,16. En los pacientes con síndrome de Down se ha observado una prevalencia de EPS del 1 %, y parece existir una predisposición a desarrollar lesiones más generalizadas, y más resistentes al tratamiento16,17. Una explicación a este fenómeno es que estos pacientes sufren un envejecimiento prematuro de la piel, lo que indica cierto grado de displasia del tejido conjuntivo17. Sin embargo, también se han encontrado publicaciones que reflejan la aparición de lesiones localizadas en estos pacientes8.
Los hallazgos histopatológicos son característicos, e incluyen un aumento de las fibras elásticas en la dermis papilar, gruesas y homogéneas, que tienden a eliminarse a través de la epidermis por unos conductos rellenos de fibras elásticas fragmentadas y detritos nucleares basófilos. Las fibras elásticas pierden sus propiedades tintoriales al entrar en el canal, que suele estar recubierto por un tapón de queratina. La epidermis aparece hiperplásica y acantósica, adoptando un aspecto en garra, intentando «engullir» las fibras elásticas anormales. Pueden encontrarse escasas células inflamatorias y células gigantes de cuerpo extraño, en la dermis próxima de este canal. En la región central, el tejido granulomatoso se reemplaza por un tejido fibroso sin fibras elásticas, formando clínicamente una cicatriz atrófica superficial.
En el microscopio electrónico se observan las fibras elásticas aumentadas, gruesas, onduladas, ramificadas y más refringentes que las fibras elásticas normales18.
Múltiples terapias han demostrado ser efectivas en el tratamiento de la EPS, aunque ninguna está considerada como el tratamiento de elección. Incluyen la crioterapia, los corticoides tópicos e intralesionales, y los agentes queratolíticos. Otras alternativas descritas son el tazaroteno, láser de dióxido de carbono, láser de colorante pulsado de 585 nm y el empleo de retinoides orales en las formas generalizadas. Debe evitarse la cirugía, el electrocauterio, y la dermoabrasión, por el riesgo de formación de queloides en estos pacientes7,19,20.


