





Dermatofibrosarcoma protuberans is the most common skin sarcoma, although its incidence is very low compared with other skin tumors. It presents as a slow-growing indurated plaque on which nodules develop over time. The lesion arises in the dermis but can invade subcutaneous tissue, fascia, muscle and even bone. COL1A1-PDGFB translocation is specific to dermatofibrosarcoma protuberans, and the presence of this fusion contributes to diagnosis in certain cases. A review of the literature provides evidence that recurrence is much lower after Mohs micorgraphic surgery than after conventional wide local excision. In the case of metastatic disease or when surgery would be mutilating, another recently approved treatment is the tyrosine kinase inhibitor imatinib.
El dermatofibrosarcoma protuberans es el sarcoma de piel más frecuente aunque su incidencia es muy baja comparada con otros tumores cutáneos. Se presenta clínicamente en forma de placa indurada de crecimiento lento sobre la que aparecen nódulos a medida que el tumor progresa. Se localiza inicialmente en la dermis desde donde infiltra el tejido celular subcutáneo, la fascia, el músculo e incluso el hueso. La translocación COL1A1-PDGFB es específica del dermatofibrosarcoma protuberans y sirve de ayuda en el diagnóstico de determinados casos. Según la revisión de las series publicadas en la literatura, el porcentaje de recidivas con cirugía micrográfica de Mohs es mucho menor que el encontrado cuando se emplea cirugía convencional con márgenes amplios. Para casos metastásicos o en aquellos donde la cirugía pueda ser mutilante se dispone recientemente del imatinib, fármaco de la familia de los inhibitores de la tirosina quinasa.
Dermatofibrosarcoma protuberans (DFSP) is a cutaneous tumor representative of the advances in diagnosis and treatment in oncology gained through an understanding of molecular biology. Certain cases can be diagnosed through the presence of a specific translocation and there is a promising protein tyrosine kinase inhibitor that has opened up the possibility of treatment of advanced disease.
DFSP was first described in 1890 by Taylor1 as a sarcoma resembling a keloid. Darier and Ferrand2 were the first to recognize DFSP as a unique entity in 1924. One year later, Hoffman3 coined the terms Darier-Ferrand tumor or dermatofibrosarcoma protuberans in reference to the particular tendency of this tumor to form protruding nodules on the skin.
DFSP is currently defined as a slow-growing infiltrative skin tumor with a high rate of local recurrence but low metastatic capacity. According to the World Health Organization,4 DFSP is classified as a fibrous, fibrohistiocytic, or histiocytic tumor. Weiss and Goldblum,5 in the book Enzinger and Weiss's Soft Tissue Tumors, considered it as a fibrohistiocytic tumor of intermediate malignancy.
Although DFPS has traditionally been classified as a fibrohistiocytic tumor, the histogenesis of DFSP remains uncertain. According to different studies, the origin of DFSP may be fibroblastic,6,7 histiocytic,6,8 or neural.9,10 CD34+ dermal dendrocytes have been proposed as another possible origin.11,12 However, many of these studies have contradictory results and none clearly demonstrate which type of cell DFSP is derived from.
The expression in DFSP of nestin, an intermediate filament expressed on neuroectodermal stem cells, suggests that DFSP originates from pluripotent neuromesenchymal stem cells.13–15 This hypothesis, which considers the origin of certain tumors to be a mutated pluripotent stem cell, is currently the most widely accepted for DFSP. This type of nestin-positive mesenchymal stem cell is found in the hair follicle.14
As for most sarcomas, DFSP does not have a well-established risk factor and its etiology is unknown. In 1951, Pack and Tabah16 suggested that local trauma in the region of the tumor was an etiologic factor on the basis that such an event was reported by 13% of the patients in their series. Subsequently, Taylor and Helwig17 found that local trauma had occurred in 19 out of a series of 115 cases (16.5%). Since then, there have been numerous reports of DFSP in a region affected by trauma. A history of trauma, which is present in 10% to 20% of cases, could trigger the appearance of the tumor or just be a coincidence.
Several cases of DFSP have been reported in women in whom tumor growth starts or accelerates during pregnancy.18 In fact, extensive expression of progesterone was found in 3 cases of DFSP in pregnant women,19 and attempts have been made to link this finding with a possible hormonal etiology of DFSP, although results so far have been inconclusive. What does seem clear is that tumor onset is not related to sun exposure as no studies report such an association.
EpidemiologyDFSP is an uncommon tumor, with an estimated incidence of between 0.8 and 5 cases per million inhabitants per year.20–22 The annual incidence seems to be greater in blacks than in other races,22,23 and it appears to affect men and women equally.17,24,25 Although extensive series of cases show a higher incidence in men than women, in the review of 2885 cases performed by Criscione and Weinstock,22 there was a slightly higher incidence in women and in the series of 143 patients studied by Martín et al.,26 63% of the patients were women.
For all races and both sexes, incidence peaks between 30 and 50 years,22 although congenital cases and cases in elderly individuals have been reported.
DFSP is most frequently located on the trunk, as reflected by all the large series in which 40% to 60% of the cases appear at this site,16,17,22,26,27 particularly on the shoulder girdle and back. The second most frequent site is the limbs, which account for 20% to 30% of cases. The head and neck are involved in 10% to 15% of cases; tumors on this site typically present on the scalp and the supraclavicular region.16,17,22,26,27
Clinical CharacteristicsDFSP usually presents as a small plaque with a flesh, brownish, pinkish, or even violaceous coloration. In this initial stage, it might go unnoticed by the patient and often be confused with a benign tumor, given that it is an asymptomatic, nonspecific lesion. The tumor grows slowly in this initial plaque stage. Three different appearances are possible.26 The first is morphea-like, in which the lesion appears as an indurated plaque with a flesh, whitish, or grayish color. In the second, the atrophoderma-type tumor presents as a soft, depressed flesh-colored plaque with an atrophic appearance. The third, an angioma-type tumor, is less common and resembles vascular lesions such as flat angioma. As the tumor grows, it infiltrates more deeply and spreads, and nodules start to develop on the surface (Fig. 1A–D). The time taken for the transition from the plaque phase or nonprotruding phase to the nodular phase is highly variable, with a range of less than 1 month to up to 50 years.26,27

Clinical images of dermatofibrosarcoma. A, Dermatofibrosarcoma in the left frontal region in the form of a plaque. B, Dermatofibrosarcoma in the supraclavicular region with surface nodules. C, Dermatofibrosarcoma on the chest with the appearance of a scarred plaque. D, Dermatofibrosarcoma in the supraclavicular region with surface nodules.
The size of the tumor depends essentially on the duration of growth. Normally, when the tumor is seen in the clinic, it usually has a size of 1 to 5cm,27 but sizes of greater than 20cm have been reported.16
The tumor is usually located in the dermis and infiltrates the subcutaneous cellular tissue, and so it is usually mobile with no fixation to deeper structures, although long-standing tumors can invade the fascia, muscle, periosteum, and bone.16,23,27,28
Histopathologic CharacteristicsMacroscopically, DFSP appears as a single, fairly well-delimited mass in the dermis. It has a firm consistency and a yellowish or gray color. In the macroscopic examination, infiltration of subcutaneous cell tissue is usually evident (Fig. 2A). Microscopically, DFSP appears as a well-differentiated fibrosarcoma in the histologic study. It is composed of a dense and uniform proliferation of spindle cells, with large and elongated nuclei, negligible pleomorphism, and low mitotic activity.23,27 The stroma has variable quantities of collagen and capillaries. One of the most important histologic characteristics of DFSP is the arrangement of these cells in intermingled bundles in an irregular or storiform pattern23,27 (Fig. 2B). In a less common presentation, the cells may be arranged radially around a central fibrous hub in a cartwheel pattern.
. B, Storiform pattern in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100). C, Infiltration of dermatofibrosarcoma in the form of digitform structure (hematoxylin and eosin, original magnification ×40). D, Muscular infiltration in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×40).")
Histologic images of dermatofibrosarcoma. A, Histologic image of dermatofibrosarcoma, low-magnification view (hematoxylin and eosin, original magnification ×12.5). B, Storiform pattern in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100). C, Infiltration of dermatofibrosarcoma in the form of digitform structure (hematoxylin and eosin, original magnification ×40). D, Muscular infiltration in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×40).
The epidermis over the tumor is usually thinned and has flattened epidermal ridges. There is usually no involvement of the papillary dermis, and a Grenz zone or transition region between the epidermis and the tumor is present.17
The cell density is much higher in the central part of the tumor than in the peripheral regions, where long, thin extensions can be seen in the form of fibrous bundles with low cellularity that infiltrate the subcutaneous cellular tissue, muscular fascia, and muscle, and extend down to the bone17,23,27 (Fig. 2C and D). These tentacle-like extensions, which can reach far from the central part of the tumor, mean that subclinical extension of DFSP is highly unpredictable. The structures may go unnoticed in a conventional histologic study, causing a high rate of recurrence, even after excision with wide margins.29,30 It has been calculated that the microscopic extension of the tumor ranges from 0.3 to 12cm beyond the macroscopic borders.29
The essential characteristic of DFSP is the way it infiltrates the subcutaneous cell tissue. Infiltration usually occurs along the septa, and even along the lobes, to give a honeycomb appearance (Fig. 3A). In 1990, Kamino and Jacobson31 differentiated between the infiltrative patterns of DFSP and dermatofibroma. They found that DFSP, in addition to the honeycomb pattern, infiltrated the subcutaneous cellular tissue most often in a multilayer pattern or in bands parallel to the skin surface, leaving uninvolved strips of fat within the tumor. Likewise, they saw that 60% of the cases of DFSP infiltrated in a parallel-band pattern, 30% infiltrated in a honeycomb pattern, and 10% shared the 2 patterns. Subsequently, Zelger et al.32 confirmed the existence of 2 patterns of infiltration for DFSP, although the honeycomb pattern was more common in their series, in contrast to the one of Kamino and Jacobson.
. B, Fibrosarcomatous pattern in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100). C, Staining with CD34 in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100).")
Histologic images of dermatofibrosarcoma. A, Honeycomb infiltration of subcutaneous cell tissue (hematoxylin and eosin, original magnification ×40). B, Fibrosarcomatous pattern in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100). C, Staining with CD34 in dermatofibrosarcoma (hematoxylin and eosin, original magnification ×100).
DFSP was considered a tumor exclusive to adult patients until 1957, when it was reported in a 5-year-old boy. Since then, more than 30 cases of congenital DFSP have been reported in addition to many more cases in children.33–35
Congenital DFSP has the same immunohistochemical characteristics and the same molecular abnormalities as conventional DFSP, although the clinical and histologic differences are usually evident. Congenital DFSP usually presents as a macule or atrophic plaque rather than as a tumor and is very often confused with vascular malformations or tumors, morphea, atrophoderma, atrophic scarring, or cutis aplasia congenita.33,35
From the histologic point of view, early-stage tumors may lack some of the characteristic findings of DFSP such as the storiform pattern or infiltration of the subcutaneous cell tissue. In these cases, diagnosis is often delayed.
Giant-Cell FibroblastomaIn 1982, Shmookler and Enzinger36 reported a series of 20 cases of a rare fibroblastic tumor. It mainly appeared in patients aged less than 10 years and was characterized histologically by fibroblast cell proliferation along with multinucleated giant-cell proliferation in a fibromyxoid stroma. The authors named the entity giant-cell fibroblastoma. Seven years later, the same authors proposed that giant-cell fibroblastoma be considered an infantile variant of DFSP.37 The close relationship between giant-cell fibroblastoma and DFSP was shown through the presence of the same clinical, histologic, and immunohistochemical characteristics and the same molecular abnormalities.38 However, it differs from conventional DFSP in that the age of onset is more often younger than 10 years, giant multinucleated cells are present, and there is a myxoid stroma in histology.
Pigmented Dermatofibrosarcoma Protuberans (Bednar Tumor)In 1957, Bednar39 described a tumor, supposedly of neural origin, that showed a storiform pattern along with pigmented spindle cells. The author named this entity pigmented storiform neurofibroma. Subsequently, it was noted that Bednar tumor shared clinical and histologic characteristics with DFSP, and so the tumor is currently considered a pigmented variant.27 Bednar tumor accounts for 1% to 5% of cases of DFSP and appears more often in black patients, unlike conventional DFSP.27,40 Clinically, it is usually indistinguishable from conventional DFSP although it may appear pigmented if there is sufficient melanin in the tumor. Pigmentation is, however, usually a histologic finding. Indeed, histologically, the essential characteristic of Bednar tumor is the presence of a population of dendritic cells with melanin in a greater or lesser proportion. From the immunohistochemical point of view, as with conventional DFSP, the tumor is positive for CD34 and negative for S100.27,41
Cases of DFSP that recurred in the form of Bednar tumor42 and also the presence of fibrosarcomatous areas in metastatic Bednar tumor have been reported. In addition, Bednar tumor has specific chromosomal abnormalities that distinguish it from conventional DFSP.43 This all illustrates the common prognosis and nature of the 2 types of tumor.
Atrophic Dermatofibrosarcoma ProtuberansIn 1985, Lambert et al.44 reported 5 cases of DFSP with clinical characteristics resembling a plaque morphea or a morpheaform basal cell carcinoma, given that the surface appeared depressed. The term dermatofibrosarcoma nonprotruberans was proposed because, histologically, the lesions were clearly cases of DFSP. Cases similar to those of Lambert and coworkers were later published, and the term atrophic DFSP came to be used to refer to DFSP with the clinical appearance of morphea.
Clinically, the entity consists of an irregular plaque, occasionally depressed, with an atrophic appearance, sometimes with superficial telangiectasias, and a flesh, erythematous, or brownish coloration.45
Atrophic DFSP is defined histologically by a decrease of more than 50% in the thickness of the dermis compared to healthy peritumoral dermis.46 Cases of atrophic DFSP with the characteristic DFSP translocation have been reported.47
Sclerosing Dermatofibrosarcoma ProtuberansSclerosis within the tumor is rare. When it does occur, it appears to be a spontaneous process, with no trigger or history of inflammatory processes or radiotherapy. Of the 72 cases of DFSP reported by Díaz-Cascajo et al.48 only 2 (5%) had sclerotic changes in more than 50% of the tumor mass, thereby confirming that sclerosis is uncommon in DFSP.
From the histologic point of view, in the sclerotic areas, the neoplastic cells are gradually replaced by sclerotic tissue without a notable decrease in the tumor thickness, as is the case with atrophic DFSP. The amount of collagen in these areas correlates with the loss of tumor cellularity.49 Cases have also been reported in which the sclerotic areas correspond to tumor nodules.50 The sclerotic areas are partially positive for CD34, and positivity correlates with a decrease in tumor cells, which remain embedded in the sclerotic stroma.
For Díaz-Cascajo et al,48 excessive production of collagen by tumor cells, without concurrent inflammation, and the presence of transition between typical areas of DFSP and sclerotic areas may be a sign of involution. The 3 cases described by Hattori50 correspond to long-standing DFSP, and could provide support for the theory of Díaz-Cascajo and coworkers.
Myoid Dermatofibrosarcoma ProtuberansThe presence of areas of myoid differentiation in DFSP is a rare occurrence that was first described in 1996 by Calonje and Fletcher.51 These authors reported 5 cases of common DFSP or DFSP with a fibrosarcomatous component. Disperse bundles and nodules were present with confluent eosinophilic spindle cells, a well-defined cytoplasm, and vesicular nuclei very similar to those of smooth muscle cells or myofibroblasts. These myoid areas were negative for CD34 but did stain for actin. The areas appear both on the surface and in the deep layers of the tumor. They do not tend to be located close to the muscle, arrector pili, or vascular walls, but seem to be randomly distributed, indicating that they formed their own areas of the tumor. However, many authors think that these myoid areas do not reflect true differentiation of DFSP but rather a reactive phenomenon that presents in the form of hyperplastic stromal myofibroblasts.52,53
Regardless of the exact significance of this finding, the presence of areas of myoid differentiation in DFSP is rare and usually occurs in cases of DFSP with a fibrosarcomatous component.51
Myxoid Dermatofibrosarcoma ProtuberansThe myxoid variant of DFSP was first described in 1983 by Frierson and Cooper.54 Subsequently, several case reports have been published as well as an extensive series of 23 cases by Reimann et al.55 in 2007. All cases coincide in highlighting that myxoid DFSP is very uncommon.
From the clinical and prognostic point of view, the tumor is similar to conventional DFSP, with the exception that it appears to present more frequently on the limbs than on the trunk.55 Histologically, the tumor is composed of spindle cells or stellate cells with a palely eosinophilic cytoplasm in a lobulated arrangement. In the stroma, the abundant presence of myxoid material, which by definition has to be present in more than 50% of the tumor,55 means that the storiform pattern is less noticeable. Mitosis is limited and there is usually little pleomorphism. Characteristically, there are numerous branched vessels with thin walls. CD34+ cells can be detected in most cases. This finding and the honeycomb infiltration pattern are usually the strongest diagnostic criteria given that in most cases histologic diagnosis is complex.
Dermatofibrosarcoma Protuberans With a Fibrosarcomatous ComponentIn 1951, Penner reported a case of metastatic DFSP which contained areas of fibrosarcoma.56 In the following years, many cases and series of DFSP with a fibrosarcomatous component (DFSP-FS) have been published. Wang et al.57 performed a study in which they separately analyzed the DFSP area and the fibrosarcomatous area in 6 patients with DFSP-FS. They found the same molecular abnormalities in all 6 samples of DFSP and in 5 samples of fibrosarcomatous tissue, providing support for the common histogenesis of these 2 components. The presence of fibrosarcomatous areas in the DFSP is observed in 7% to 17% of cases according to the series.40,58,59 From the clinical and epidemiological point of view, DFSP-FS is no different from conventional DFSP. Histologically, there are areas indistinguishable from a fibrosarcoma within the DFSP lesion. These areas are characterized by a dense proliferation of spindle cells, arranged in long bundles that cross to form a herringbone pattern.58 The fibrosarcomatous areas can occupy from 5% to 90% of the tumor, but according to Weiss and Goldblum,5 DFSP is considered to have a fibrosarcomatous component when the occupation is at least 5% to 10% of the tumor (Fig. 3B). Immunohistochemical staining with CD34 in the fibrosarcomatous areas is usually less intense or absent compared to contiguous areas of DFSP. In contrast, it has been shown that there is overexpression of p5359 and a high mitotic index in areas of fibrosarcoma.
Immunohistochemical CharacteristicsIntermediate FilamentsVimentin is expressed in all mesenchymal cells; thus almost all sarcomas, including DFSP, are positive for this protein.60
Nestin is an intermediate filament that was first described as a marker of neuroectodermal stem cells and subsequently identified in mesenchymal stem cells in bone marrow, lung, muscle, and the pancreas.11 Recently, the usefulness of this protein has been demonstrated in DFSP, particularly to differentiate it from dermatofibroma (DF). Mori et al.,15 compared staining with nestin and other immunohistochemical markers in 16 cases of DFSP and in 30 cases of DF. Positive results were obtained for nestin in 94% of DFSP samples and in only 13% of DF samples. In other more recent studies, even more robust results were obtained, with intense staining being reported in all cases of DFSP.13,14
CD34In normal skin, CD34 is expressed in endothelial cells, in perifollicular spindle cells, in perivascular and periadnexal spindle cells, and in interstitial dendritic cells of the reticular dermis.61,62
CD34 is considered a relatively specific marker for vascular neoplasms, but in 1992 Aiba et al.62 studied CD34 expression in a series of fibrohistiocytic tumors such as DF, DFSP, hypertrophic and keloid scarring, and found that the only tumor that expressed it was DFSP. Similar findings were later reported by Kutzner.63 In 1994, Cohen et al.64 reviewed all studies published until then (9 series involving 96 cases) and calculated that 88% of DFSP are CD34 positive. Since then, several studies have reported results of immunostaining with CD34, with positive results in 92% to 100% of cases65–67; it therefore seems to be a very useful marker for differentiating between DFSP and other fibrohistiocytic tumors, especially DF (Fig. 3C).
Even though most cases of DFSP express CD34, positive staining for CD34 has been reported in many benign and malignant tumors, such as inflammatory myofibroblastic tumor, solitary fibrous tumor, sclerotic fibroma, superficial acral fibromyxoma, Kaposi sarcoma, neurofibroma, perineurioma, and melanoma.61
Factor XIIIaFactor XIIIa is a tetrameric protein that is expressed in normal skin and in dermal dendrocytes of the papillary dermis, especially around superficial vessels.67 In 1989, Cerio et al.68 reported positive staining for factor XIIIa in 30 cases of DF, whereas in 16 cases of DFSP, staining was absent or limited, suggesting that this marker could be useful for differentiating between these 2 tumors. Most studies report positivity in between 90% and 95% of DF and, in addition, expression occurs in most cells and is very intense, unlike in DFSP, where staining is positive only in 10% to 15% of cases.64,67,69
Other Immunohistochemical MarkersDifferential diagnosis of DFSP is often established with DF and so, CD34 (positive in most cases of DFSP and in up to 25% of DF69) and factor XIIIa (negative in most cases of DFSP and positive in almost all cases of DF) are the most widely used markers; however other markers have also been reported to be useful.
In 2004, West et al.70 studied the expression of apolipoprotein D (ApoD) in 421 soft tissue tumors and found that the tumor with most extensive expression of this marker was DFSP, unlike common DF, which were all negative. Subsequently, other studies confirmed that ApoD may be interesting for differentiating between DFSP and DF.
Another immunohistochemical marker that has been studied in DFSP is stromelysin 3. In the studies of this marker, the results show more extensive expression in DF than in DFSP, with the differences being more or less striking depending on the study.71
Most studies are in agreement that DFSP has a low proliferative index as determined both with Ki-67 and MIB-1.59,72
Immunohistochemical expression of p53 is usually negative or weakly positive in DFSP and most frequently associated with DFSP-FS.59 However, p53 may be useful for distinguishing between DFSP and DF given that this protein is not usually expressed in DF.
Cytogenetics and Molecular Biology of Dermatofibrosarcoma ProtuberansThe cytogenetics of DFSP was first described in 1990 by Bridge et al.73 and Mandahl et al.74 with 2 cases of DFSP in which a supernumerary ring chromosome of indeterminate origin was found. Subsequently, fluorescence in situ hybridization (FISH) showed that the DFSP ring contained sequences of chromosome 17.75 The combination of FISH techniques and comparative genomic hybridization showed the involvement of chromosome 22 in the formation of the ring chromosome.76 Since then, the combination of sequences from chromosome 17 and 22 in a supernumerary ring chromosome derived from chromosome 22 has been considered a characteristic feature of DFSP.43
Simon et al.77 with FISH and molecular biology techniques, identified the exact fusion point and found 2 genes present (platelet-derived growth factor beta [PDGFB] [22q13.1] and the collagen I alpha 1 gene [COL1A1 [17q.22]) giving rise to a chimeric gene.
In all published cases of the COL1A1-PDGFB fusion gene in DFSP, the exact cleavage site for the PDFGB gene is constant at exon 2; however, the cleavage site for the COL1A1 gene is very variable. The literature describes 27 different exons as implicated, with exons 24, 29, and 32 being the most frequently involved, although the cleavage site for COL1A1 does not seem to have any impact on clinical characteristics, histology, or prognosis.58
Different studies suggest that the result of translocation with the COL1A1-PDGFB fusion gene in DFSP leads to a chimeric protein that is subsequently processed to give rise to mature and fully functional PDGFB.
Therefore, t(17;22) in DFSP is associated with activation of the PDGFB receptor through autocrine and paracrine production of a functional ligand that translates into a chronic mitogenic signal, able to induce neoplastic transformation.
To date, DFSP is the only tumor in which a somatic abnormality in the COL1A1 and PDFGB genes has been demonstrated. In addition to translocation t(17;22) seen in DFSP, other numeric and structural chromosomal abnormalities have been described in DFSP. Of these, trisomy 8 is the most frequent abnormality as it is present in a third of karyotyped DFSP lesions.76
Radiologic CharacteristicsDSFP has been studied with conventional radiography, computed tomography, and even with arteriography, but none of these techniques have provided precise information and there are no specific characteristics of these test results in DFSP that may help in diagnosis or better define its location.
According to the largest magnetic resonance imaging (MRI) study in DFSP,78 this technique is more sensitive and specific than clinical palpation for determining the depth of infiltration; however, the reliability of MRI is lower in tumors located on the head and neck.
Treatment of Dermatofibrosarcoma ProtuberansFull excision is the treatment of choice for DFSP.27,29 It should be taken into account that the growth pattern of DFSP, with long, thin structures, makes the tumor very asymmetric with a subclinical extension reaching a long way from the center.29 These tentacle-like structures at the periphery of the tumor may go unnoticed, even in a conventional histologic study, explaining the high rate of local recurrence in DFSP.23
There is still debate about whether conventional surgery or Mohs micrographic surgery (MMS) is preferable for the treatment of DFSP.
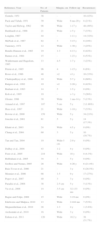
Conventional Surgery for Dermatofibrosarcoma ProtuberansThere are many studies in the literature of DFSP treated with conventional surgery. However, the approach to tumor excision varies greatly and so it is difficult to draw specific conclusions about the use of conventional surgery in DFSP. Table 1 shows the series of at least 10 cases of DFSP treated with conventional surgery. From 1951 to 2011, there were 38 such series, which included a total of 1782 patients.16,17,20,24,25,30,79–110 On analyzing the resection margin, most series used the term wide margin, without specifying exactly how much healthy skin was excised; moreover, in many cases, the tumors were excised several times until histologically negative margins were achieved.102,105,109 Other series used a safety margin ranging from 1 to 5cm. There is a trend towards a lower number of relapses in cases excised with larger safety margins.88,101 Many of the studies do not report whether or not underlying muscular fascia were spared during the excision of DFSP. Nevertheless, it can be calculated that DFSP recurs in up to 60% of cases of excision with wide margins, without specifying either the margin used or whether or not muscular fascia were included in the excision. Only 9 of the 38 series did not report any recurrences and in more than half of these series, recurrences were reported in more than 20% of cases.
Series With 10 or More Cases of Dermatofibrosarcoma Treated With Conventional Surgery.
| Reference, Year | No. of Patients | Margin, cm | Follow-up | Recurrences |
| Gentele, 1951 | 38 | - | - | 16 (42%) |
| Pack and Tabah, 1951 | 39 | Wide | 6 mo-20 y | 8 (21%) |
| Taylor and Helwig, 1962 | 98 | Wide | 1-17 y | 48 (49%) |
| Burkhardt et al., 1966 | 21 | Wide | > 5 y | 7 (33%) |
| Longhin, 1967 | 44 | - | 1-11 y | 14 (32%) |
| McPeak et al., 1967 | 82 | 3 | 3-15 y | 8 (10%) |
| Tamoney, 1971 | 12 | Wide | 1-30 y | 3 (25%) |
| Bendix-Hansen et al., 1983 | 19 | 1-3 | 4-13 y | 8 (42%) |
| Barnes et al., 1984 | 15 | - | 1-23 y | 8 (53%) |
| Waldermann and Hagedorn, 1985 | 13 | 4-5 | 1-7 y | 3 (23%) |
| Petoin et al., 1985 | 96 | 4 | 1-15 y | 6 (6%) |
| Roses et al., 1986 | 48 | >2 | >3 y | 16 (33%) |
| Chattopadhyay et al., 1986 | 10 | Wide | 5-7 y | 6 (60%) |
| Rutgers et al., 1992 | 19 | >2 | 2-28 y | 8 (42%) |
| Brabant et al., 1993 | 14 | 5 | 1-5 y | 0 (0%) |
| Koh et al., 1995 | 19 | - | > 3 y | 5 (26%) |
| Gloster, 1996 | 39 | Wide | 1 mo-14 y | 5 (13%) |
| Arnaud et al., 1997 | 107 | 5cm | 5 y | 2 (1.86%) |
| Hass et al., 1997 | 21 | Wide | 1-10 y | 7 (33%) |
| Bowne et al., 2000 | 159 | Wide | 5 y | 34 (21%) |
| Joucdar et al., 2001 | 81 | 5 | 5 y | 14 (17.3%) |
| Khatri et al., 2003 | 24 | Wide | 4.5 y | 0 (0%) |
| Chang et al., 2004 | 60 | 3 | 5 y | 10 (16.7%) |
| Tan and Tan, 2004 | 10 | Wide-3cm | 2-9 y | 0 (0%) |
| DuBay et al., 2004 | 43 | 1-2 | 4 y | 0 (0%) |
| Fiore et al., 2005 | 218 | Wide | 10 y | 8 (4.3%) |
| Behbahani et al., 2005 | 34 | 3 | 5 y | 0 (0%) |
| Szollosi and Nemes, 2005 | 28 | Wide | 4-26 y | 6 (21.4%) |
| Ruiz-Tovar et al., 2006 | 21 | - | 3 y | 6 (28.5%) |
| Monnier et al., 2006 | 66 | 1-5 | 9 y | 17 (27%) |
| Popov et al., 2007 | 40 | 3 | 3 y | 0 (0%) |
| Paradisi et al., 2008 | 38 | 2-5cm | 5 y | 5 (13%) |
| Yu et al., 2008 | 14 | 3-5cm | 32-133 mo | 0 (0%) |
| Bague and Folpe, 2008 | 15 | Wide | 3-19 mo | 0 (0%) |
| Edelweiss and Malpica, 2010 | 13 | Wide | 2-444 mo | 7 (53%) |
| Meguerditchian et al., 2010 | 28 | 1-3 | 4 y | 1 (3.6%) |
| Archontaki et al., 2010 | 16 | Wide | 3 y | 0 (0%) |
| Erdem et al., 2011 | 120 | Wide | 10.2 y | 38 (31.7%) |
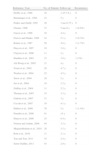
The first author to report use the use of MMS was Dr Mohs himself in 1978.111 Since then, many individual case reports and series have reflected the usefulness of this technique in the treatment of DFSP and propose it as the treatment of choice.112 Twenty-six series with 10 cases or more of DFSP treated with MMS have been published (Table 2).29,30,78,96,105,109,113–133 The recurrence rates reported with MMS in the 27 series of DFSP range from 0% to 8.3%, that is, much lower than the rates reported with conventional surgery with wide margins. In addition, 22 of the 27 series did not report any recurrence with follow-up in excess of 2 years. In addition, MMS provides the true, narrow margin of healthy tissue in each case, unlike conventional surgery with wide margins, which may be incomplete or include a large quantity of healthy, uninvolved tissue.
Series With 10 or More Cases of Dermatofibrosarcoma Treated With Mohs Micrographic Surgery.
| Reference, Year | No. of Patients | Follow-up | Recurrences |
| Hobbs et al., 1988 | 10 | 1.25-7.6 y | 0 |
| Breuninger et al., 1994 | 23 | 5 y | 0 |
| Parker and Zitelli, 1995 | 20 | 3 mo-8.75 y | 0 |
| Gloster, 1996 | 15 | 5 mo-8 y | 1 (6.6%) |
| Garcia et al., 1996 | 16 | 4.4 y | 0 |
| Dawes and Hanke, 1996 | 24 | 5.1 y | 2 (8.3%) |
| Ratner et al., 1997 | 58 | 4.8 y | 1 (1.7%) |
| Haycox et al., 1997 | 10 | 3.4 y | 0 |
| Clayton et al., 2000 | 11 | 2 y | 0 |
| Huether et al., 2001 | 33 | 3.8 y | 1 (3%) |
| Ah-Weng et al., 2002 | 21 | 4 y | 0 |
| Nouri et al., 2002 | 20 | 4.7 y | 0 |
| Wacker et al., 2004 | 22 | 4.5 y | 0 |
| Snow et al., 2004 | 29 | 5 y | 0 |
| Sei et al., 2004 | 10 | 2.2 y | 0 |
| DuBay et al., 2004 | 11 | 5.2 y | 0 |
| Thomas et al., 2007 | 35 | 3.3 y | 0 |
| Gattoni et al., 2007 | 31 | 3 y | 0 |
| Cecchi et al., 2007 | 10 | 4.1 y | 0 |
| Häfner et al., 2008 | 70 | 5 y | 1 (1.4%) |
| Paradisi et al., 2008 | 41 | >5 y | 0 |
| Hancox et al., 2008 | 25 | 8.4 y | 0 |
| Nelson and Arlette, 2008 | 44 | 3.3 y | 0 |
| Meguerditchian et al., 2010 | 20 | 3.3 y | 0 |
| Roh et al., 2010 | 11 | 2.1 y | 0 |
| Tan and Tan, 2011 | 35 | 2.4 y | 0 |
| Serra-Guillén, 2011 | 43 | 2.9 y | 0 |
The most widely used technique is the variant known as the slow Mohs procedure.125 This is performed as follows (Fig. 4A–D). First, there is a debulking or excision of the tumor or scar tissue itself in the case of recently excised tumors with involved margins. Then, the first stage of 0.5 to 1cm of clinically healthy skin is taken, using a scalpel angle of 45° and cutting down through the entire subcutaneous tissue and the most superficial layers of the muscular fascia. Before removing this first stage, the specimen is mapped with silk sutures and a photograph taken. The debulked material is processed conventionally, fixed in formol, and embedded in paraffin. Parallel cuts are then stained with hematoxylin and eosin. The first Mohs stage is pinned to a plaque of polyurethane foam to ensure that it retains its shape and to prevent retraction. The sample is sent in a recipient containing formol. The photograph of the specimen before removal from the patient is also sent to the pathologist. The pathologist first separates the lateral margins with cuts from the epidermis to the bottom of the specimen. The base is then detached with horizontal cuts. All pieces obtained are assigned a number and the whole process is reproduced from the photograph. The cuts are stained with hematoxylin and eosin and studied under the microscope, looking for evidence of DFSP. In the event that doubtful areas or those difficult to interpret are found, immunohistochemical staining of the same section is undertaken with CD34. If, after studying all the margins, no evidence of DFSP is found, the surgical defect is closed. If, however, a margin with evidence of disease is found, excision continues, with referrence to the photograph to determine exactly the involved margin. This procedure is repeated until all margins are negative. Once this has occurred, the defect is closed definitively. The advantage of the slow Mohs technique is that the cuts are of much greater quality and it is much easier to find evidence of DFSP, unlike conventional Mohs techniques which, because the specimen analyzed is frozen, provide a histologic section that is much harder to interpret. The slow Mohs technique, however, takes much longer than the conventional Mohs technique.

Slow Mohs surgery for dermatofibrosarcoma protuberans. A, Debulking or narrow-margin excision of the tumor. B, First stage of the Mohs procedure. Mapped ring of clinically healthy skin. C, Surgical defect after first stage. Exposure of the muscle plane. D, Division of the specimen by the pathologist.
Identification of unregulated expression of the PDGDB receptor as a result of translocation t(17;22) led to the hypothesis that inhibitors such as imatinib of the protein tyrosine kinase present in the receptor could be active in DFSP. Imatinib binds competitively to the PDGF receptor and blocks its tyrosine kinase activity. After encouraging results in preclinical studies, several clinical studies have reported good response to imatinib in metastatic and locally advanced DFSP.108,134–153 The agent has been used as neoadjuvant, prior to excision, with responses ranging from reductions in tumor size of 19% to complete clinical response (Table 3).108,134–153 However, the studies in which complete response have been obtained should be reviewed critically before more widespread use of imatinib in DFSP. With regard to the 4 cases with complete response published by McArther et al.,138 in addition to the short follow-up period, histologic study was only performed in 2 patients. After treatment with imatinib, the histologic findings characteristically show a tumor with low cellularity and abundant hyalinized collagen, which can be falsely interpreted as disease-free tissue.138,141 Therefore, the COL1A1-PDGFB translocation should be studied to demonstrate or rule out tumor persistence after treatment with imatinib in DFSP that has apparently regressed in conventional histology. Nevertheless, according to all studies published, imatinib appears to be beneficial as neoadjuvant treatment in cases of locally advanced disease or as palliative treatment in cases of metastatic disease.138,141
Published Cases of Dermatofibrosarcoma Protuberans Treated With Imatinib.
| Reference, Year | Case | Duration of Treatment | Response | Follow-up |
| Rubin et al., 2002 | Metastatic | 4 mo | Partial (reduction 75%) | - |
| Maki et al., 2002 | Metastatic | 4 wk | Partial | Death |
| Metastatic | 2 mo | Almost complete | ||
| Mizutani et al., 2004 | Metastatic | 3 mo | Partial | - |
| Labropoulos et al., 2005 | Metastatic | 20 mo | Complete | 20 mo |
| McArthur et al., 2005 | Locally advanced | 698 d | Partial | 845 d |
| Locally advanced | 62 d | Partial | 699 d | |
| Locally advanced | 141 d | Partial | 572 d | |
| Locally advanced | 457 d | Complete | 536 d | |
| Locally advanced | 139 d | Partial | 258 d | |
| Locally advanced | 188 d | Complete | 267 d | |
| Locally advanced | 146 d | Complete | 225 d | |
| Locally advanced | 88 d | Complete | 88 d | |
| Metastatic | 198 d | Partial | 383 d | |
| Metastatic | 21 d | Stable | 32 d | |
| Price et al., 2005 | Locally advanced | 23 wk | Partial | - |
| Mehrany et al., 2006 | Locally advanced | 24 mo | Partial | 18 mo |
| Savoia et al., 2006 | Locally advanced | 11 mo | Partial | 6 mo |
| Kasper et al., 2006 | Metastatic | 4 mo | Partial | - |
| Wright et al., 2007 | Locally advanced | 5 mo | Partial | 16 mo |
| Kim et al., 2007 | Metastatic | - | Complete | 1 y |
| Heinrich et al., 2008 | 12 cases (unspecified) | 3-12 mo | 4 complete6 partial1 progression1 unknown | 23.9 mo |
| Thomison et al., 2008 | Locally advanced | 1 y | Partial | - |
| Lemm et al., 2008 | Locally advanced | 3 mo | Partial (reduction 60%) | 6 mo |
| Llombart et al., 2009 | Metastatic | 4 mo | Partial | Death |
| Locally advanced | 1 y | Partial (reduction 50%) | 1 y | |
| Han et al., 2009 | Locally advanced | 3 mo | Partial (reduction 19%) | 1.5-4 y |
| Locally advanced | 3.5 mo | Partial (reduction 45%) | ||
| Locally advanced | 7 mo | Partial (reduction 62%) | ||
| Locally advanced | 3 mo | Partial (reduction 22%) | ||
| Rutkowski et al., 2010 | 24 cases7 “metastatic” | 248-328 d | 9 partial8 stable4 progression3 not evaluable | 2.6 y |
| Gooskens et al., 2010 | Locally advanced | 6 mo | Partial (reducción 30%) | 3.5 y |
| Locally advanced | 12 mo | Partial | 3 y | |
| Locally advanced | 6 mo | Partial | 6 mo | |
| Kerob et al., 2010 | 25 cases20 “primary”5 “recurrences” | 2 mo | PartialReduction < 30% in 16 cases> 30% in 9 cases | - |
| Edelweiss and Malpica, 2010 | Locally advanced | - | - | 72 mo |
| Stacchiotti et al., 2011 | Locally advanced | 4 mo | Partial | 5 mo |
| Metastatic | 4 mo | Partial | Death | |
| Metastatic | 4 mo | Partial | Death | |
| Metastatic | 4 mo | Partial | Death | |
| Rutkowski et al., 2011 | 15 cases9 “locally advanced”6 “metastatic” | - | 10 partial2 stable3 progression | 16 mo |
The role of radiotherapy in the management of DFSP has not been extensively studied and has generated some debate. Given that surgical treatment can guarantee cure in most cases, radiotherapy does not seem appropriate as there is insufficient experience of its use in DFSP.
Most published cases and series correspond to DFSP excised with narrow or positive margins, with the subsequent use of radiotherapy.92,154 Thus, radiotherapy is reserved for truly inoperable disease, when imatinib treatment is not possible, or as palliative treatment.
PrognosisThe most recognized factor for poor prognosis in DFSP is inappropriate excision with positive resection margins or positive areas very close the surgical border.5,27,94 Such situations are directly related to the possibility of recurrence and to progression of DFSP to highly differentiated fibrosarcomatous-type histology.
Clinically, large tumors and those located on the head and neck appear to have a worse prognosis.155
Histologically, the presence of areas of fibrosarcoma within DFSP seem to be associated with a more aggressive course,25,59,156 as does an increased mitotic index, a greater cell density,25 and p53 mutation.59
Lymph node involvement in DFSP is very uncommon. Very few isolated cases have been reported of DFSP with metastasis to lymph nodes. According to the review by Rutgers et al.,24 only 11 regional lymph node metastases were reported in a series of 913 cases, that is, 1%. Metastasis, however, was associated with a much worse prognosis; when metastasis occurred, most of the patients died within 2 years of the appearance of lymph node involvement. Lymph node metastasis is up to 3 times less frequent than visceral metastasis.5 Routine lymph node resection is therefore not recommended in DFSP.
In most cases of uncomplicated DFSP, the appearance of visceral metastasis is an exceptional event, generally thought to occur in between 1% and 5% of cases157; however, these data are based on old series where perhaps some patients received inadequate treatment and the risk of metastasis was higher.157 In addition, with the introduction of imatinib, there have been many recent reports of metastatic DFSP that would not have been published had it not been for the treatment with this agent. It is therefore very difficult to correctly calculate the rate of metastasis in DFSP.
What is well known is that most metastatic disease occurs when DFSP has recurred several times or histology reveals areas of fibrosarcoma.24,25,59 The most frequent site for metastasis in DFSP is the lung,151,153 accounting for up to 75% of cases of metastasis.5
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Serra-Guillén C, et al. Dermatofibrosarcoma protuberans. Actas Dermosifiliogr. 2012;103:762-77.


