






Introducción
El dermatofibrosarcoma protuberans (DFSP) es una neoplasia de partes blandas de origen cutáneo poco frecuente y caracterizada por un comportamiento biológico de malignidad intermedia. Aunque produce metástasis sólo excepcionalmente, cursa con gran morbilidad por su gran capacidad infiltrativa local y la elevada tasa de recidivas tras la extirpación quirúrgica. Las descripciones iniciales de esta entidad fueron realizadas por Darier y Ferrand en 1924, a la que denominaron dermatofibroma progresivo y recurrente1. Un año más tarde, y atendiendo a la tendencia de este tumor a desarrollar nódulos protuberantes, Hoffman acuñó el término de dermatofibrosarcoma protuberans2. La mayor parte de las descripciones iniciales del DFSP se realizaron teniendo en cuenta sus características clínicas y su tendencia a recidivar tras la exéresis quirúrgica. En 1962, Taylor y Helwig, en una primera revisión de 150 casos, describen con detalle las características histológicas de la lesión, caracterizando la proliferación fibroblástica de aspecto poco sarcomatoso de células tumorales organizadas en fascículos agrupados en forma de remolinos o en rueda de carro3.
En 1992 se realizaron las primeras descripciones de la positividad inmunohistoquímica del DFSP para el CD344, que sigue siendo el principal apoyo diagnóstico inmunohistoquímico de este tumor, especialmente si se asocia la negatividad de la mayor parte de casos de DFSP para el factor XIIIa5. En 1997 se demostró que muchos DFSP muestran, de forma característica, la traslocación t (17;22)6. La región de ruptura en esta traslocación t (17;22) afecta normalmente al exón 2 del gen del factor de crecimiento derivado de plaquetas beta (PDGFB), localizado en el cromosoma 22, que se fusiona con el gen del la cadena alfa 1 del colágeno I (COL1A1) localizado en el cromosoma 17. En todos los casos el gen COL1A1 se expresa con gran intensidad, actuando como inductor de la transcripción génica. El resultado de esta fusión es la elaboración de una proteína de fusión COL1A1-PDGFB, que se procesa a nivel extracelular hasta transformarse en PDGFB completamente maduro y funcional. El PDGF liberado es capaz de inducir un estímulo mitógeno mediante la activación de su receptor. Por lo tanto, el producto de fusión COL1A1-PDGFB induce una activación del receptor PDGFBR, mediante la producción autocrina y paracrina de su ligando funcional.
En los últimos años se ha avanzado mucho en el conocimiento de la citogenética de este tumor, en el reconocimiento de sus características inmunohistoquímicas y se han introducido técnicas específicas en el diagnóstico, estadiaje y tratamiento que permiten un control adecuado de la enfermedad en la mayoría de los pacientes. Este artículo pretende revisar y actualizar los datos conocidos acerca de la incidencia, epidemiología, hallazgos histológicos, inmunohistoquímicos, citogenéticos, pronósticos y terapéuticos de este tumor, con especial énfasis en las descripciones de la biología molecular de este tumor y las alternativas terapéuticas de la cirugía de Mohs y de los inhibidores del PDGFr.
Epidemiología
El DFSP es un tumor poco frecuente. Su incidencia en EE.UU. se ha calculado entre 0,8 y 4,5 casos/millón de habitantes/año7-9. Una reciente revisión epidemiológica recoge una incidencia anual de 4,2 casos por millón de habitantes/año10. Supone un 0,1 % de todos los cánceres y un 1 % de todos los sarcomas de partes blandas11. Sin embargo, si se tienen en cuenta exclusivamente los tumores cutáneos, el DFSP es el sarcoma de origen cutáneo más frecuente.
El DFSP se ha descrito en todas las razas. Se calcula que la incidencia de DFSP en la raza negra es aproximadamente el doble que en la raza blanca, sin que se tenga una explicación para esta diferencia10. La variedad pigmentada del DFSP (tumor de Bednar), se da también predominantemente en pacientes de raza negra12.
La mayor parte de series largas de pacientes muestran una distribución por sexos homogénea, o con ligera preferencia por varones. Rutgers et al en una revisión de la literatura sobre 902 casos encontraron una relación varón/mujer (V:M) de 3:213. Sin embargo, más recientemente en una revisión epidemiológica de Criscione et al sobre 2.885 casos recogidos en las bases de datos del NCI se recoge una preferencia por el sexo femenino10.
El DFSP puede aparecer a cualquier edad, aunque es mucho más frecuente entre los 20 y 50 años. Se han descrito desde casos congénitos, hasta casos en pacientes mayores de 90 años14,15. Existen pocos casos de DFSP descritos en edad pediátrica, aunque al tratarse de un tumor asintomático que suele diagnosticarse tardíamente por su lento crecimiento, muchos casos diagnosticados en edad adulta se han iniciado en edad pediátrica7. La proporción de casos pediátricos en las series publicadas de DFSP oscila entre el 6 y el 20 %3,16,17. Debe tenerse en cuenta que actualmente se considera al fibroblastoma de células gigantes (FCG) como la variedad juvenil de dermatofibrosarcoma. El FCG fue descrito inicialmente en 198218 como un tumor propio de la infancia, caracterizado histológicamente por una proliferación fusocelular de distribución estoriforme con áreas mixoides y por la presencia típica de células gigantes multinucleadas que rodean espacios pseudovasculares. Desde su descripción, ambas entidades se habían clasificado independientemente, hasta que en 1989, Shmookler et al describieron varios casos de FCG con hallazgos clínicos e histológicos propios de DFSP19. Esta relación entre ambos tumores ha sido posteriormente confirmada por la descripción de numerosos casos que comparten hallazgos propios de DFSP y de FCG, tanto desde el punto de vista clínico como inmunohistoquímico y citogenético20. En una reciente revisión sobre cáncer cutáneo en pacientes menores de 25 años, el DFSP-FCG fue el tercero en frecuencia, con un 13 % de los casos, por detrás del melanoma y el carcinoma basocelular21.
Los antecedentes de traumatismo como posible factor etiológico en el DFSP son objeto de controversia. Posiblemente dichos antecedentes favorezcan el desarrollo del tumor, ya que entre un 10 y un 20 % de los casos refieren historia previa de traumatismo3,11,22. Así mismo, se han descrito casos de DFSP asentado en cicatrices quirúrgicas23, postraumáticas24, quemaduras25, radiodermitis26, puntos de inyección de vacunas 27 y de puntos de inyección de vías centrales28.
El DFSP se localiza preferentemente en el tronco. Entre un 40 y un 50 % de los casos se sitúan en esa área, generalmente en el tórax y los hombros; entre un 30 y un 40 % de los casos en la porción proximal de las extremidades (es más frecuente en la extremidad superior que en la inferior) y en un 10-15 % de los casos el DFSP en cabeza y cuello, generalmente en cuero cabelludo y región supraclavicular. La localización en la extremidad cefálica comporta una mayor dificultad en el tratamiento que justifica una elevada tasa de recidivas cuando se compara con otras zonas29. Cuando ocurre en cuero cabelludo, hasta en un 25 % de los casos se produce invasión del periostio30. La localización acral del DFSP es excepcional, especialmente en adultos. Gloster, en una revisión de la literatura, señala que el DFSP de manos y pies supone tan sólo un 0,01 % de los casos11. Sin embargo, se ha señalado que el DFSP infantil tiene mayor tendencia a la localización acral. Rabinowitz et al en una revisión de 27 casos previamente publicados de DFSP en edad infantil encontraron que el 14,8 % se localizaban en manos o pies31. Martin et al en 1998 realizan una revisión de 140 casos de DFSP en edad infantil publicados previamente, y demuestran que la única diferencia del DFSP infantil con respecto a la forma del adulto es la mayor frecuencia de localización acral17. Esta tendencia en los casos infantiles de DFSP no se ha visto confirmada por una tendencia a la localización acral en las series de revisión de FCG18, considerando la forma infantil del DFSP, por lo que no se puede afirmar con rotundidad la tendencia a localización acral del DFSP en la infancia. De hecho, en una revisión de la literatura de los 150 casos pediátricos de DFSP publicados hasta la fecha, la proporción de casos de dicha localización es de menos del 9 %.
Clínica
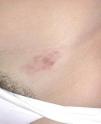
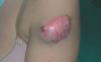
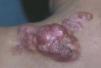
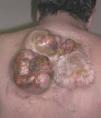
El DFSP es un tumor de crecimiento lento, indolente, por lo que los pacientes suelen consultar tardíamente. El aspecto clínico del DFSP depende del tiempo de evolución. En su inicio suele presentarse como una placa única, de carácter asintomático, de coloración violácea, roja-marronácea o rosada, de consistencia dura y adherida a la piel, pero no a planos profundos (fig. 1). Con el tiempo, la placa puede mantenerse estable durante un largo periodo, crecer lentamente o entrar en una fase de crecimiento rápido desarrollando múltiples nódulos, de ahí su nombre de protuberante (figs. 2 y 3). Excepcionalmente, el DFSP se manifiesta desde su inicio como uno o múltiples nódulos intradérmicos de color rojo púrpura y consistencia firme.
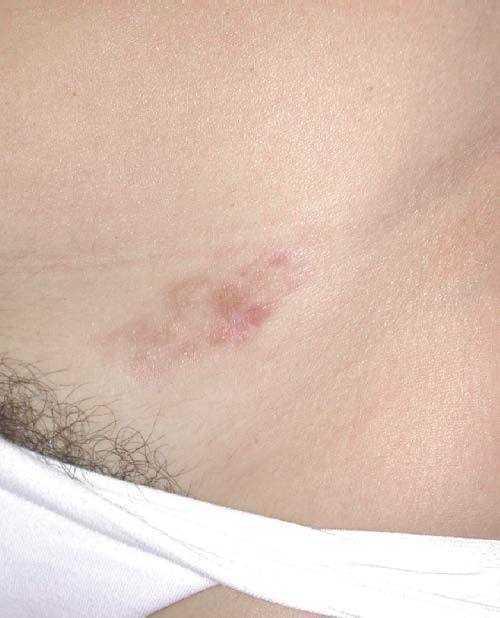
Figura 1. Dermatofibrosarcoma en fase inicial. Placa infiltrada de tonalidad marronácea con pequeños nódulos en superficie.

Figura 2. Dermatofibrosarcoma en fase proliferativa. Nódulo de rápido crecimiento en brazo.

Figura 3. Dermatofibrosarcoma protuberans. Múltiples nódulos excrecentes sobre cicatriz de extirpación previa.
En las fases iniciales del DFSP, cuando aún no ha adquirido el aspecto protuberante que caracteriza la lesión, son frecuentes los errores diagnósticos, interpretándose la lesión como cicatriz hipertrófica. Por esta razón, Martin et al han estudiado los patrones clínicos del DFSP en sus fases iniciales32. Así distinguen, dentro de la forma no protuberante de DFSP 3 formas de presentación clínica: a) forma morfea-like, caracterizada por formar una placa indurada, blanca o marrón, con aspecto de cicatriz, morfea, carcinoma basocelular morfeiforme o dermatofibroma en placa; b) forma atrofodermia-like, caracterizada por una placa de consistencia blanca y deprimida, blanca o marrón, que asemeja una atrofodermia o anetodermia, y c) forma angioma -like, la menos frecuente, compuesta por placas rojas o violáceas, induradas o blandas, que clínicamente recuerdan a lesiones vasculares tipo malformación vascular o angioma. De acuerdo con esos autores, la forma más frecuente de presentación en el adulto es la de una placa de gran tamaño con múltiples nódulos en su superficie. En niños, y sobre todo cuando las lesiones se localizan en el tronco, son más frecuentes las formas no protuberantes como placa morfea -like y los casos congénitos, como atrofodermia-like.
El tamaño de la lesión en el momento del diagnóstico suele estar entre 2 y 5 cm de diámetro, aunque con frecuencia las lesiones son ignoradas por el paciente y pueden llegar a medir 25 cm.
La lesión adopta un patrón de crecimiento infiltrativo, por lo que la palpación muestra adherencia a la piel circundante. Las lesiones de larga evolución o las recidivadas pueden mostrar infiltración de estructuras profundas como la fascia, músculo, periostio o hueso.
Histología
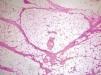
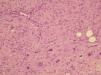
La apariencia histológica del DFSP es la de un fibrosarcoma bien diferenciado. El tumor se localiza inicialmente en la dermis y está compuesto por una densa proliferación de células fusiformes, monomorfas, de núcleo grande y elongado, generalmente con escaso pleomorfismo y una baja actividad mitótica. El estroma suele ser escaso, con presencia de depósito de colágeno intercelular y pequeños capilares. Las células fusiformes se disponen en fascículos entrelazados irregularmente, siguiendo un patrón estoriforme (fig. 4). En algunas áreas, las células se disponen radialmente en torno a un foco central de colágeno acelular, siguiendo un patrón en rueda de carro, que fue señalado por Taylor y Helwig como de gran valor diagnóstico3.

Figura 4. Visión panorámica de dermatofibrosarcoma protuberans. Proliferación fusocelular sin atipias con patrón estoriforme clásico ocupando la totalidad de la dermis. (Hematoxilina-eosina, x20.)
La principal característica histológica del DFSP es su capacidad para infiltrar los tejidos circundantes a una distancia considerable del foco central del tumor. La celularidad es mayor en la zona central que en la periferia del tumor, que muestra bordes infiltrativos hacia la dermis circundante y el subcutis. Las células tumorales infiltran el tejido celular subcutáneo, en forma de tentáculos, a través de los septos y los lobulillos (fig. 5). Estas prolongaciones del tumor son escasamente celulares y pueden asemejar tractos fibrosos normales en un examen rápido, lo que dificulta la determinación de la magnitud real de la lesión, y ser causa de recurrencias después de una resección aparentemente amplia. La afección de la fascia, los músculos subyacentes y el periostio, así como la ósea es un evento tardío (fig. 6).
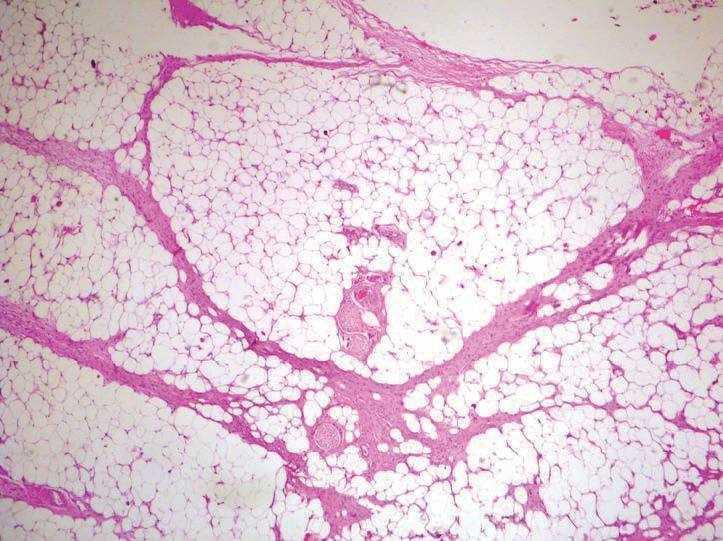
Figura 5. Extensión al tejido celular subcutáneo mediante tentáculos tumorales que se extienden a través de los septos. (Hematoxilina-eosina, x10.)
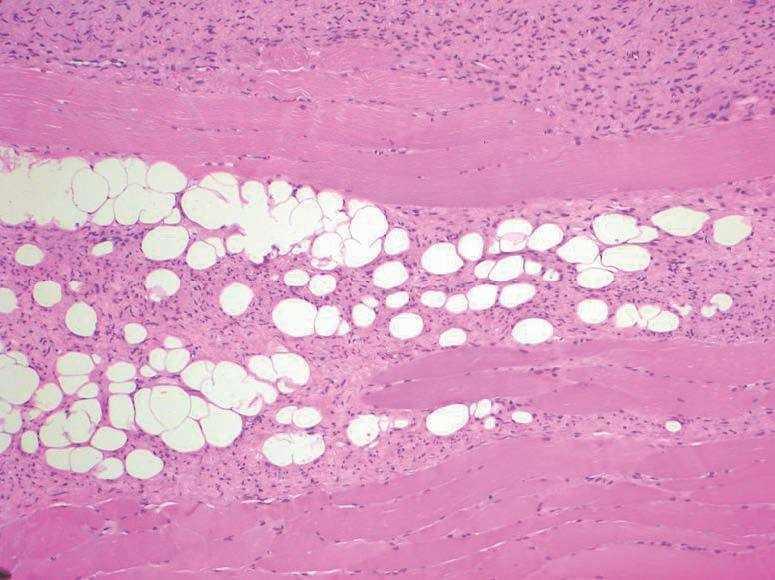
Figura 6. Infiltración de musculatura estriada por dermatofibro sarcoma protuberans. (Hematoxilina-eosina, x20.)
El diagnóstico diferencial histológico debe realizarse fundamentalmente con otras neoplasias de aspecto fibrohistiocítico, como el fibrosarcoma, histiocitoma fibroso maligno y dermatofibroma. Los fibrosarcomas son muy raros en la piel, suelen localizarse más profundamente, y están compuestos por una proliferación fusocelular de marcado pleomorfismo y elevado índice mitósico. La disposición de los fascículos celulares suele seguir un patrón en herradura o en espina de pescado más que estoriforme.
El DFSP en sus fases iniciales de placa puede asemejar un dermatofibroma de variedad celular o fibrosa. En ocasiones el dermatofibroma celular puede mostrar un patrón estoriforme e infiltrar el tejido celular subcutáneo, dificultando el diagnóstico diferencial con el DFSP. Sin embargo, el dermatofibroma cuando infiltra la grasa lo hace bien con un patrón radial siguiendo los septos del subcutis o bien con un patrón compresivo en profundidad. Por el contrario, el DFSP infiltra la grasa en un 30 % de los casos siguiendo un patrón en panal de abeja, mientras que en un 70 % de los casos lo hace en un patrón multicapa, con bandas fusocelulares, orientadas paralelamente a la superficie cutánea33,34.
Inmunohistoquímica
A principios de los años noventa, varios trabajos describieron que el antígeno CD34 se expresa en un 50-90 % de los DFSP, hecho que no se observó en otros tumores fibrohistiocitarios que en ocasiones pueden confundirse con un DFSP, como el dermatofibroma, el histiocitoma fibroso maligno, la miofibromatosis infantil, el fibrosarcoma, la cicatriz hipertrófica o el queloide35,36. Desde su introducción en la clínica diaria, se ha considerado que la expresión del CD34 es característica y fundamental para el diagnóstico diferencial del DFSP. Sin embargo, con el tiempo se ha ido observando que otros sarcomas también pueden expresar CD34, como el sarcoma miofibroblástico inflamatorio (fibrosarcoma inflamatorio), el miofibrosarcoma, el angiosarcoma o el sarcoma epitelioide; por lo tanto, actualmente se debe considerar al CD34 como menos específico de DFSP. Incluso, en los últimos años, diversos trabajos han demostrado que algunas lesiones fibrohistiocitarias benignas como el tumor fibroso solitario37, el fibroma esclerótico38, el fibromixoma superficial acral39, los fibromas digitales celulares40, el fibroma de la nuca41 o incluso algunos dermatofibromas42 también pueden expresar este marcador.
En el diagnóstico diferencial entre DFSP e histiocitomas fibrosos celulares resulta de gran utilidad el factor XIIIa, que suele ser negativo en los DFSP. Sin embargo, no todos los histiocitomas fibrosos expresan XIIIa; por esta razón en los últimos años se han descrito nuevos paneles inmunohistoquímicos para el diagnóstico diferencial entre ambas entidades, como la estromelisina III, la apolipoproteína D (APO D), y el CD16342-44. Estos tres marcadores, al igual que el factor XIIIa suelen ser positivos en dermatofibromas y negativos en el en DFSP.
Variedades histológicas
Existen diferentes variedades histológicas del DFSP, algunas de ellas, como la fibrosarcomatosa, condicionan un peor pronóstico, y su presencia debe ser valorada en el estudio histológico.
Dermatofibrosarcoma mixoide
Es frecuente que el DFSP contenga áreas pequeñas de degeneración mixoide. Sin embargo, en ocasiones, el patrón histológico predominante en la totalidad del tumor es mixomatoso. El DFSP mixoide se caracteriza por la presencia de áreas moderadamente celulares constituidas por células estrelladas o fusiformes con cúmulos abundantes de mucina hialuronidasa-sensible en el espacio intercelular. La trama vascular del tumor suele ser muy abundante, y pueden encontrarse áreas de DFSP clásico en el espesor de la lesión, densamente celulares y con patrón estoriforme. El DFSP mixoide no tiene ninguna característica clínica diferente a la del DFSP convencional, ni un pronóstico distinto, por lo que su reconocimiento es útil sólo para el diagnóstico diferencial con otros sarcomas mixoides como el fibrosarcoma mixoide, el mixofibrosarcoma inflamatorio o el liposarcoma mixoide.
Dermatofibrosarcoma pigmentado (tumor de Bednar)
Fue descrito por Bednar en 1957. Es un tumor fusocelular y estoriforme que contiene una población en cantidad variable de células dendríticas que fabrican melanina. Posiblemente esta población provenga de melanocitos dérmicos que colonizan el tumor. Dependiendo de la cantidad de melanina que contenga la lesión, ésta puede llegar a ser clínicamente pigmentada, aunque en ocasiones es simplemente un hallazgo microscópico. Se calcula que un 1 % de los DFSP son pigmentados.
Dermatofibrosarcoma fibrosarcomatoso
La variante fibrosarcomatosa del DFSP se considera una lesión poco frecuente. No se conoce con exactitud la incidencia de cambios fibrosarcomatosos en el DFSP, pero se ha situado entre el 3 y el 10 % de todos los DFSP dependiendo de las series45,46. Se caracteriza histológicamente por una proliferación densa fusocelular, agrupada en fascículos que se disponen siguiendo un patrón en herradura o en espina de pescado. La celularidad es abundante, muestra atipia moderada y un índice mitósico elevado (fig. 7). Acompañando las zonas fibrosarcomatosas, se pueden encontrar áreas de DFSP convencional, sin atipias. La proporción de áreas fibrosarcomatosas oscila entre el 30 y 50 % de la totalidad del tumor. Debe tenerse en cuenta que estas áreas transformadas no expresan, o lo hacen débilmente, el CD34, lo que constituye una dificultad diagnóstica añadida. Esta variedad histológica se caracteriza por un comportamiento biológico más agresivo, con mayor tasa de recidivas locales tras la cirugía y un mayor riesgo de metástasis a distancia47. Se ha señalado que esta transformación en el DFSP está favorecida por las recidivas previas en el tumor después de cirugías insuficientes46, aunque este extremo no se ha confirmado en todas las series45,47. La aparición de áreas fibrosarcomatosas en el DFSP no es más que una progresión de grado histológico en el sarcoma, posiblemente debida a la aparición de nuevas alteraciones genéticas en el tumor, como la presencia de nuevas copias de la traslocación COL1A1-PDGFB48 o mutaciones en la p5347,49, que favorecen un comportamiento biológico más agresivo, con tumores que crecen mucho más rápido, llegando a originar metástasis con frecuencia (fig. 8).
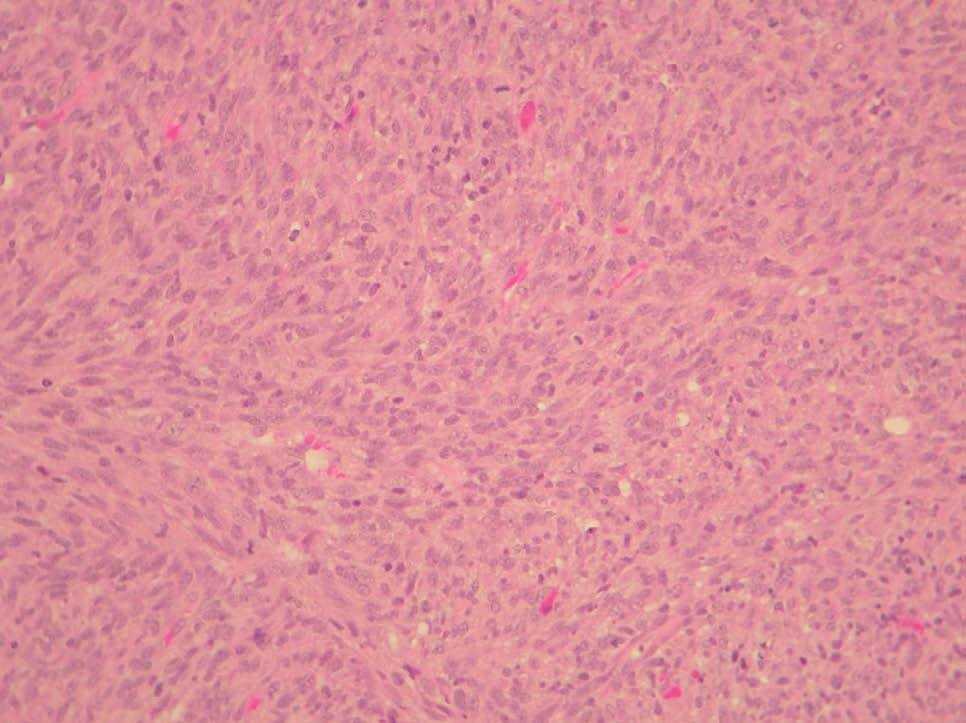
Figura 7. Dermatofibrosarcoma protuberans con áreas fibrosarcomatosas. Proliferación fusocelular en fascículos siguiendo patrón en espina de pescado, con abundantes mitosis y atipias nucleares. (Hematoxilina-eosina, x20.)
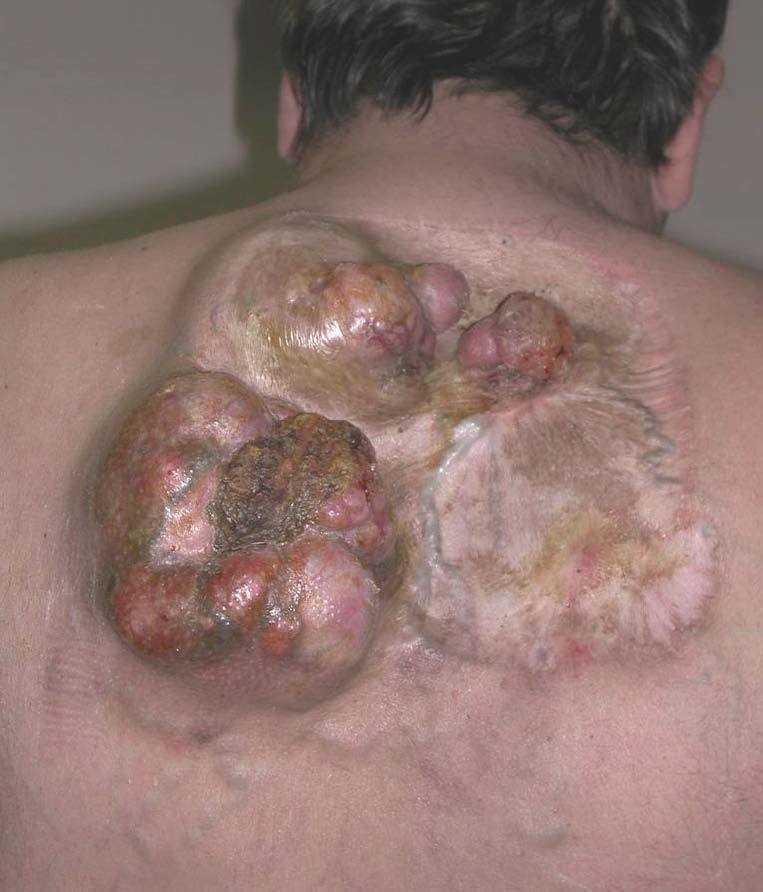
Figura 8. Dermatofibrosarcoma protuberans con transformación fibrosarcomatosa. Gran tumor recidivado en espalda sobre cicatrices de extirpaciones previas.
Dermatofibrosarcoma atrófico
Es una variante histológica caracterizada por una marcada atrofia en la dermis suprayacente al tumor, que reduce su espesor a menos de la mitad. Clínicamente se caracteriza por la aparición de una placa deprimida, de consistencia generalmente blanda, que se diagnostica clínicamente de anetodermia, atrofodermia o morfea. Se han publicado unos 30 casos, la mayor parte de ellos localizados en el tronco, aunque en la edad infantil lo hacen preferentemente en extremidades50. El DFSP atrófico no tiene un pronóstico diferente al del resto de DFSP convencionales, y posiblemente sea debido a la secreción por parte de las células tumorales de fragmentos de COL1A1 de una longitud determinada que induzca la digestión de la dermis por parte de colagenasas51.
Dermatofibrosarcoma con áreas de fibroblastoma de células gigantes
Ya se ha comentado anteriormente que el FCG es una variedad de DFSP propia de la infancia. No obstante, algunos casos de DFSP del adulto presentan zonas en el tumor con células gigantes rodeando espacios sinusoidales que caracterizan al FCG (fig. 9).
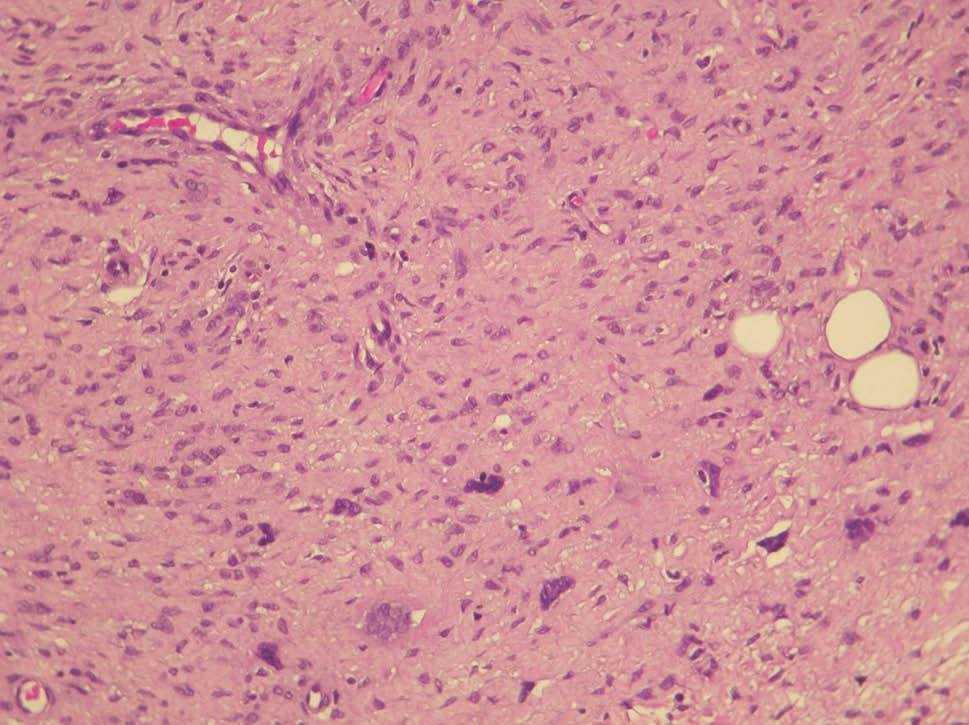
Figura 9. Dermatofibrosarcoma protuberans con áreas de fibroblastoma de células gigantes. (Hematoxilina-eosina, x20.)
Otras variedades histológicas de DFSP
Además de las comentadas anteriormente, se han descrito diferentes variedades histopatológicas del DFSP, cuyo reconocimiento tiene valor para el diagnóstico diferencial con otros tumores, pero que no comportan un valor pronóstico distinto al del DFSP convencional. Estas variedades son el DFSP esclerótico52, el DFSP con áreas mioides53 y el DFSP con células granulares54.
Citogenética y biología molecular del DFSP
Desde 1990 se conoce la existencia ocasional de cromosomas en anillo supernumerarios en algunos DFSP55. Posteriormente, el análisis de hibridación in situ con fluorescencia (FISH) mostró que el anillo del DFSP contenía secuencias del cromosoma 1756. La combinación de las técnicas del FISH y la hibridación genómica comparada (CGH) mostraron también la implicación del cromosoma 22 en la formación del cromosoma en anillo, conteniendo bajos niveles de amplificación de las regiones 17q22-qter y 22q10-q13.1. Desde entonces, se considera característica del dermatofibrosarcoma la presencia de cromosomas en anillo derivados del cromosoma 22 conteniendo secuencias del cromosoma 17 y 2257. Estos cromosomas en anillo pueden encontrarse también en casos de fibroblastoma de células gigantes. En 1995, se publicó por primera vez la presencia de la traslocación t(17;22) en DFSP58. Posteriormente se describieron 4 nuevos casos de DFSP, 3 en niños y un adulto, encontrando varias formas de t(17;22) en los cromosomas en anillo59. Los estudios citogenéticos y de FISH determinaron que en la mayoría de DFSP los cromosomas en anillo y lineares der(22) contienen secuencias del cromosoma 22 entre el centrómero y la banda 22q13.1 y también secuencias del cromosoma 17 entre 17q21/22 y 17 qter. La combinación de FISH y la biología molecular han ayudado a identificar la fusión del PDGFB en 22q13.1 con COL1A1 (17q/22)6.
En estos estudios se ha demostrado que más de un 90 % de los casos de DFSP muestran la traslocación t(17;22), sin embargo, se ha tratado siempre de series cortas de pacientes, por lo que la frecuencia real de la traslocación está todavía por demostrar. La región de ruptura en la traslocación t(17;22) afecta normalmente al exon 2 del gen del factor de crecimiento derivado de plaquetas beta (PDGFB), localizado en el cromosoma 22, que se fusiona con COL1A1 localizado en el cromosoma 17. En este último se han descrito diferentes puntos de ruptura que afectan a diferentes exones del gen. En todos los casos el gen COL1A1 se expresa con gran intensidad, actuando como inductor de la transcripción génica60.
La proteína de fusión COL1A1-PDGFB es procesada a nivel extracelular tras la transcripción hasta transformarse en PDGFB completamente maduro y funcional, capaz de inducir un estímulo mitógeno mediante la activación de su receptor61. Por lo tanto, el producto de fusión COL1A1-PDGFB induce una activación del receptor PDGFBR, mediante la producción autocrina y paracrina de su ligando funcional. Este receptor está constituido básicamente por tres dominios estructurales: un dominio extracelular, de unión al ligando; un dominio transmembrana de activación, y un dominio intracitoplasmático con actividad tirosincinasa que desencadena en respuesta a la unión al ligando una cascada de señalización celular sobre procesos celulares como la proliferación, quimiotaxis o apoptosis.
La identificación de la expresión aberrante del PDGFB en el DFSP como consecuencia de la traslocación t(17;22) sugiere que el uso del inhibidores específicos de tirosincinasas, como el imatinib, pueda ser útil en casos de DFSP no susceptibles de tratamiento quirúrgico radical, ofreciendo una alternativa terapéutica. De hecho, en casos aislados en los que se ha empleado imatinib en DFSP, se han obtenido elevadas tasas de respuesta, lo que sostiene la hipótesis de que las células del DFSP dependen de la expresión aberrante del PDGFRB para la proliferación y supervivencia celular62.
La presencia del gen de fusión COL1A1-PDGFB en el DFSP puede demostrarse mediante técnicas de biología molecular como es la transcriptasa inversa-reacción en cadena de la polimerasa (TI-PCR) a partir de ARN extraído de las muestras tumorales.
Este procedimiento es altamente sensible y específico y constituiría una herramienta muy útil en el diagnóstico diferencial de los DFSP con otras entidades tumorales de histología similar (fig. 10).

Figura 10. Secuenciación de la traslocación COL1A1-PDGFB.
Los problemas que genera el estudio de la traslocación mediante TI-PCR en muestras en parafina pueden solventarse con la aplicación de técnicas de FIHS, mediante sondas específicas para los dos genes implicados en la lesión, siendo ésta una técnica aplicable con más facilidad a muestras incluidas en parafina.
Evolución, tratamiento y pronóstico
El DFSP es un tumor localmente agresivo, caracterizado por baja tasa de metástasis y elevada infiltración local, por lo que el tratamiento de elección es el quirúrgico. Entre un 85-90 % de los DFSP son lesiones histológicas de bajo grado de malignidad que se caracterizan por un crecimiento infiltrativo lento, con poca tendencia a las metástasis a distancia, pero con una elevada capacidad de destrucción local y alta frecuencia de recidiva tras el tratamiento quirúrgico. Sin embargo, en un 10-15 % de los casos el DFSP puede sufrir transformaciones fibrosarcomatosas, lo que aumenta su agresividad biológica; es más frecuente en los casos de recurrencias múltiples, por lo que conseguir la resección completa de la lesión desde un principio es importante. No obstante, lograr el control local del tumor es difícil, ya que el DFSP recurre tras cirugía convencional hasta en un 30 % de los casos. Esta elevada tasa de recidivas se explica por el crecimiento excéntrico del tumor cuando infiltra el tejido celular subcutáneo. A este nivel el tumor invade en forma de tentáculos a distancia del foco inicial que pasan clínicamente desapercibidos, y que, si no se realiza un estudio histológico de márgenes muy exhaustivo, puede quedar oculto.
La realización de una escisión amplia disminuye de forma notable el índice de recidivas63. Así, cuando el margen de escisión es de 3 cm o más, la tasa de recurrencias es del 20 %, mientras que si es de menos de 2 cm aumenta a un 40 %64. Aquellas series en las que se emplean márgenes de 5 cm recogen tasas de recurrencia de menos del 5 %65.
Las recurrencias locales generalmente aparecen en los primeros 3 años tras la cirugía inicial, aunque también se han descrito casos después de muchos años, lo cual indicaría la conveniencia de realizar seguimientos a largo plazo. El principal inconveniente de la cirugía convencional con margen amplio es el de la gran morbilidad que comporta, con la extirpación de áreas amplias de tejido sano y con el riesgo de dejar tumor viable al pasar desapercibidas las prolongaciones excéntricas subcutáneas del tumor.
La cirugía micrográfica de Mohs permite el estudio completo de márgenes y ahorrar al máximo el tejido sano, por lo que se ha convertido en la técnica quirúrgica de elección en el tratamiento del DFSP. Es la técnica que consigue un mayor índice de curaciones, con una tasa de recurrencias menor del 5 %66. En el DFSP suele emplearse una modificación de la técnica de Mohs, con tejido fijado en formol e incluido en parafina (fig. 11). Este procedimiento retrasa la realización de la técnica, pero permite un mejor diagnóstico de la invasión de la grasa por el tumor, que con cortes en congelación podría pasar desapercibida67.
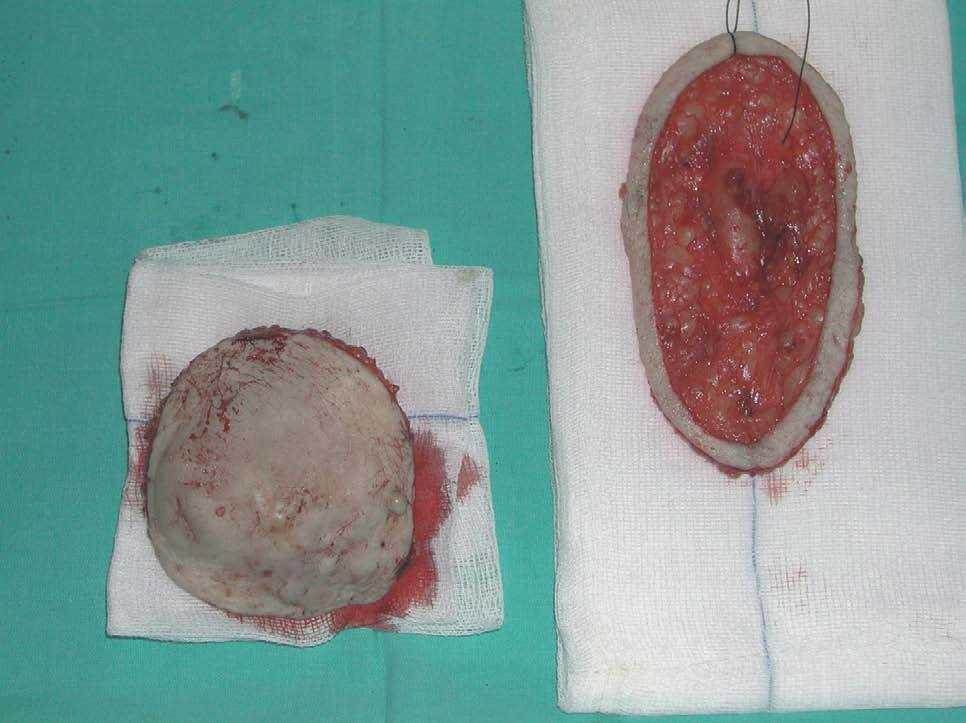
Figura 11. Realización de cirugía de Mohs diferida en un dermatofibrosarcoma protuberans. Eliminación del tumor detectable (debulking) y obtención de un estadio para estudio del margen mediante cortes horizontales en parafina.
El DFSP es un tumor que raramente produce metástasis a distancia, y esto ocurre solamente tras muchos años de evolución. La incidencia de metástasis es de un 1-3 %, con un intervalo medio desde la primera escisión de 6 años. Como en otros sarcomas, las metástasis se localizan preferentemente en el pulmón, aunque también se las ha descrito en el cerebro, el hueso y el corazón. Si bien es difícil determinar cuáles son los casos de riesgo metastático, se trata en general de lesiones recurrentes, de muchos años de evolución y con componente de fibrosarcoma en la histología.
En cuanto a las exploraciones complementarias indicadas en la evaluación de los casos de DFSP, debe tenerse en cuenta que raramente existe diseminación linfática o hematógena, por lo que sólo es necesario realizar una historia clínica completa, con estudio por aparatos, y una exploración física de los pacientes que incluya la palpación de ganglios linfáticos. La única prueba diagnóstica que parece estar indicada sería la resonancia magnética (RM) de la zona afectada, pues aporta información sobre el grado de invasión tumoral, particularmente en los casos con recurrencias frecuentes. La tomografía computarizada (TC) no está indicada, exceptuando los casos en que se sospecha que el hueso esté afectado, cuando haya sospecha de metástasis pulmonares en los casos de DFSP recurrente o muy grandes.
Nuevos tratamientos farmacológicos
El tratamiento de elección de los DFSP es la cirugía de Mohs o, en su defecto, la cirugía con margen amplio (> 3 cm). Sin embargo, no siempre es posible el tratamiento quirúrgico. El pronóstico de los casos metastásicos es muy pobre, con menos de 2 años de supervivencia tras la detección de la enfermedad metastásica y con resistencia a los tratamientos de quimioterapia convencional. Del mismo modo, los pacientes de DFSP con tumores localmente avanzados no son susceptibles de tratamiento quirúrgico radical de inicio, por lo que se precisa tratamiento neoadyuvante previo a la cirugía para disminuir el tamaño tumoral. En este sentido, la quimioterapia y la radioterapia se han mostrado poco eficaces, por lo que se hace necesario contar con nuevas alternativas terapéuticas.
Afortunadamente, el hecho de que esta entidad tumoral tenga un rasgo molecular característico, como el gen de fusión COL1A1-PDGFB, que induce la estimulación de la actividad tirosincinasa de los receptores del PDGF-BB, hace que las vías de tratamiento farmacológico se amplíen con los inhibidores de la tirosincinasa.
Imatinib mesilato (Glivec®, Novartis) ha demostrado su eficacia en otros tumores (leucemia mieloide crónica, tumores del estroma gastrointestinal) inhibiendo la actividad de la tirosincinasa de determinadas dianas moleculares (BCR-ABL7, KIT o PDGFRB). En cultivo de tejidos de DFSP, se ha demostrado que imatinib interfiere con el PDGFB. Algunos autores proponen que este fármaco induce la apoptosis de las células tumorales, lo que podría destruir totalmente el tumor, mientras otros piensan que produce una alteración del fenotipo del DFSP que disminuye su índice de proliferación y, en consecuencia, el tamaño tumoral, pero no lo elimina totalmente.
Imatinib se ha empleado con éxito tanto en casos de DFSP metastático como en casos de DFSP localmente avanzado68. Debe tenerse en cuenta que la traslocación COL1A1-PDGFB está presente en la mayoría de los DFSP, pero no en todos. Actualmente sólo puede utilizarse imatinib como fármaco de uso compasivo en pacientes con DFSP con la traslocación demostrada, sobre todo como tratamiento neoadyuvante para disminuir el tamaño tumoral antes de la cirugía. Por tanto, se abren nuevas vías de tratamiento para el DFSP, sobre todo en algunos casos como la recurrencia local, las metástasis a distancia o en zonas donde sea imposible la resección completa del tumor por ser un área de difícil acceso quirúrgico, o en casos pediátricos, donde la cirugía sea mutilante62.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Onofre Sanmartín Jiménez.
Servicio de Dermatología.
Instituto Valenciano de Oncología.
Profesor Beltrán Báguena, 8.
46009 Valencia. España.
Correo electrónico: osanmartinj@gmail.com
Aceptado el 19 de diciembre de 2006.
*Artículo parcialmente financiado por la beca de la Generalitat Valenciana GV06/274.


