



Los queratoacantomas (QA) son lesiones normalmente esporádicas y solitarias que aparecen en los pacientes que viven en zonas con exposición al sol. La exposición crónica al sol, el imiquimod, los inhibidores BRAF y los inhibidores de la vía del «erizo» pueden ser agentes desencadenantes. Las causas genéticas de los QA tales como el síndrome de Ferguson-Smith (epiteliomas escamosos autocurativos múltiples - MSSE) y el queratoacantoma eruptivo generalizado (síndrome de Grzybowski) se caracterizan por el desarrollo de docenas de cientos de QA en la edad temprana, y no están asociadas a otros tumores cutáneos o extracutáneos1. Otras enfermedades genéticas asociadas son el xeroderma pigmentosum y el síndrome de Muir-Torre (SMT).
Nuestro objetivo fue describir las características clínico-epidemiológicas y los trastornos genéticos asociados a los pacientes jóvenes con QA en nuestros centros, y desarrollar un algoritmo diagnóstico. Describimos el caso de tres pacientes que desarrollaron QA antes de los 40 años y albergaron enfermedades genéticas desconocidas predisponentes a estas lesiones cutáneas.
El paciente 1 fue un varón de 54 años de edad sin antecedentes familiares relevantes e historia personal de exposición intensa a la luz del sol, que había tenido más de diez QA desde los 39 años. Había desarrollado lesiones centrífugas crecientes en el antebrazo izquierdo tras radioterapia por un QA solitario. Las lesiones eran clínica e histológicamente sugerentes de queratoacantoma marginatum centrifugum (fig. 1). No se observó pérdida de los marcadores inmunohistoquímicos de MMR en los tumores, tratándose exitosamente las lesiones con retinoides orales y 5-fluorouracil intralesional. Se realizó un análisis de cribado del gen TGFBR1, detectándose una variante heterozigótica c.301 T>C probablemente patogénica.
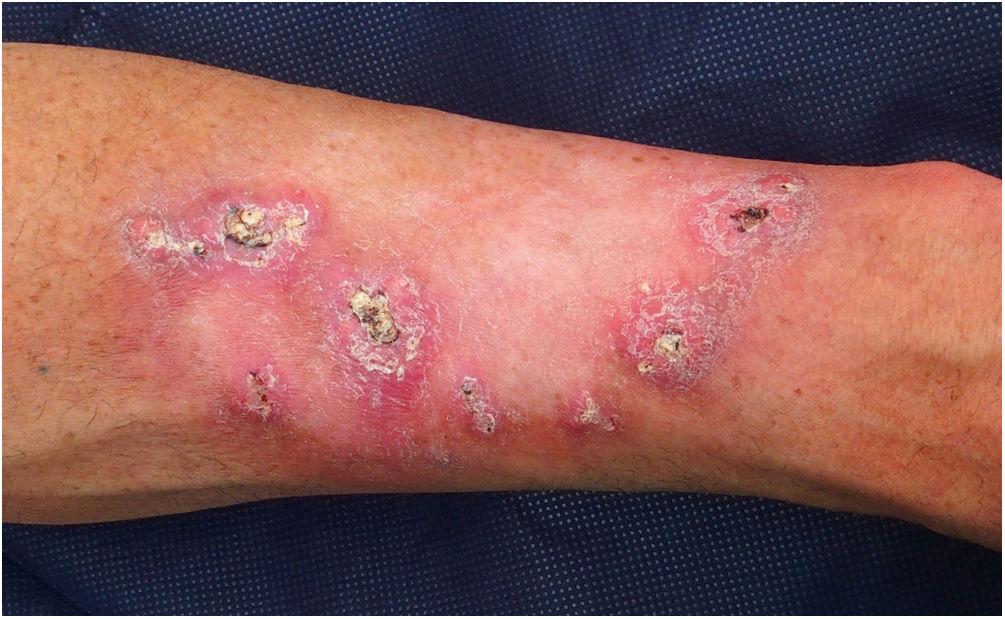
Keratoacantoma marginatum centrifugum. Nódulos queratinizantes de forma convexa de color rosado a color carne de 1 a 2 centímetros de diámetro con un tapón queratósico central, distribuidos en la periferia del campo de radiación previo (antebrazo izquierdo). El color de la figura solo puede verse en la versión electrónica del artículo.
El paciente 2 fue un varón de 38 años, de otro modo sano, que desarrolló un QA de rápido crecimiento de 13mm en la nariz. Su padre había fallecido a la edad de 53 años tras desarrollar cáncer de colon metastásico. La inmunohistoquímica de MMR reveló pérdida de MSH2 y MSH6. La lesión fue tratada con metotrexato intralesional, mostrando regresión completa en un periodo de 12 semanas. Se identificó una mutación heterozigótica constitutiva de c.142G>T en el gen MSH2 (que predijo la generación de una proteína truncada: p.Glu48*) mediante un papel de secuenciación de siguiente generación y validación de secuenciación Sanger, que confirmó el diagnóstico de síndrome de Lynch.
El paciente 3 fue un varón de otro modo sano sin antecedentes familiares, que desarrolló un QA de 17mm de diámetro en la región cervical a la edad de 38 años. Se extirpó quirúrgicamente la lesión y las tinciones inmunohistoquímicas de MMR reflejaron pérdida de MSH2 y MSH6. El estudio de genes MMR germinales mostró una gran deleción (c.(?_68)_1276+?del) que implicó a los exones 1 a 7 en el gen MSH2, lo cual confirmó el diagnóstico de síndrome de Lynch, subtipo SMT. El paciente desarrolló carcinoma pT2 de alto grado en el uréter a la edad de 41 años. También se extirparon a dicha fecha diversos adenomas sebáceos faciales y un QA en la región cervical.
El desarrollo de un QA solitario, o múltiples QA a edad temprana puede ser una señal distintiva de trastornos genéticos que pueden estar asociados a neoplasias malignas. Reconocer dichas asociaciones es importante, a fin de prevenir posibles complicaciones.
MSSE, una enfermedad autosómica dominante, está causado por las mutaciones del gen TGFBR1 junto con variantes permisivas en un locus cercano al locus TGFBR1 en el cromosoma 92. Los pacientes con MSSE desarrollan lesiones múltiples de crecimiento lento y se resuelven en meses, a medida que aparecen nuevas lesiones. Existe una penetrancia variable, que oscila entre portadores asintomáticos y pacientes que desarrollan lesiones desde la primera hasta la séptima década de la vida. Algunos pacientes pueden mostrar KCM, caracterizado por el desarrollo de lesiones de tipo arrecife de coral con expansión periférica progresiva y cicatrización central atrófica. El desarrollo de KCM tras la radioterapia debería sugerir el diagnóstico de MSSE3. Pueden utilizarse modalidades de tratamiento alternativas tales como retinoides orales, corticosteroides o metrotexato sistémico/intralesional4.
El xeroderma pigmentosum está asociado al desarrollo temprano de cáncer de piel, aunque los QA son raramente la manifestación primera del tumor. El SMT se reporta en el 10-30% de las familias con síndrome de Lynch, que albergan principalmente las mutaciones de los genes MSH2 o MLH15,6. Deberían ensayarse los marcadores inmunohistoquímicos de MMR en muestras tumorales de los pacientes con neoplasias cutáneas sebáceas, QA múltiples y/o con inicio a edad temprana. Sin embargo, la falta de expresión de las proteínas MMR en neoplasias sebáceas no es exclusiva de los pacientes con SMT, y algunos casos mantienen la expresión de las proteínas MMR con un defecto de reparación del ADN de la línea germinal subyacente.
La revisión en la literatura muestra que aproximadamente el 89% de los pacientes con QA asociados a SMT albergan mutaciones de MSH2 (tabla 1), lo cual está en concordancia con la prevalencia mutacional reportada de las neoplasias cutáneas de no QA en SMT5–11. A menudo, los QA asociados a SMT7 no se distinguen histológicamente de los QA esporádicos. Los QA en pacientes con SMT se desarrollan normalmente antes de realizarse el diagnóstico de neoplasias viscerales, mientras que los tumores sebáceos en SMT se detectan de manera común tras el diagnóstico de dichas neoplasias.
Casos reportados en la literatura de pacientes con queratoacantomas asociados al síndrome de Muir-Torre
| Autor, año | Sexo | Número de QA | Edad de la presentación (años) | Localización | Pérdida de MMR (IHC)en QA | MSI | Otras lesiones cutáneas | Tumor visceral en el índice(edad en años) | Antecedentes familiaresde neoplasias viscerales | Mutación germinal |
|---|---|---|---|---|---|---|---|---|---|---|
| Muir et al., 1967 7 | V | 6 | 32 | Cara | NR | NR | NR | -Carcinoma de células escamosas de laringe (38)-Adenocarcinoma de duodeno/íleo (40) | No | NR |
| Mangold et al., 20046 | NR | 1 | 50 | NR | MSH2 | Sí | -Adenoma sebáceo-Carcinoma de células escamosas | -Transitional cell carcinoma uréter (66)-Carcinoma de yeyuno (66) | -Cáncer de útero,cáncer de intestino delgado, cáncer abdominal (madre)-Cáncer de mama (tío materno) | MSH2 (c.1578delC) |
| Mangold et al., 20046 | NR | Múltiple | 57 | NR | NR | Sí | Tumores sebáceos múltiples | -Carcinoma urotelial (56) | -Cáncer colorrectal (hermano) | MSH2 (Deleción exones 9,10) |
| Mangold et al., 20046 | NR | 1 | 67 | NR | NR | NR | Tumores sebáceos múltiples | -Carcinomas múltiples de colon (edad desconocida) | -Cáncer colorrectal (padre, hermano) | MSH2(c.2005. 2T>C) |
| Mangold et al., 20046 | NR | Múltiple | 52 | NR | No | Sí | Hiperplasias sebáceas | - Cáncer urotelial (52).- Cáncer de vejiga (53)-Carcinomas de colon (54) | -Carcinoma de colon (padre) | MSH2 (c1571. G>C) |
| Ponti et al., 20058 | V | 4 | -34-44 | -Cuello-Tórax-Cabeza | MSH2/MSH6 | Sí | Quistes sebáceos | -Colon (35)-Pelvis renal (54) | -Vejiga-Mama | MSH2(c2131C>T) |
| Ponti et al., 20058 | M | 2 | -86 | Cabeza | -MLH1 | Sí | NR | Gástrico (89) | -Mama-Gástrico | NR |
| Stella et al., 200711 | NR | NR | NR | NR | NR | NR | NR | Colon | NR | MSH2 (deleción exones 1-6) |
| South et al., 20085 | M | 1 | 39 | NR | -MLH1 | NR | -Adenoma sebáceo | Colon (46) | NR | MLH1 (c.1489delC) |
| South et al., 20085 | M | 1 | 57 | NR | NR | NR | NR | Ninguno | Colon (hija) | MSH2 (c942. 3A>T) |
| South et al., 20085 | M | 1 | 41 | NR | NR | NR | NR | Colon (41) | NR | MSH2 (c942. 3A>T) |
| South et al., 20085 | M | 1 | 63 | NR | NR | NR | NR | -Endometrial (64)-Colon (71) | NR | MSH2 (c942. 3A>T) |
| South et al., 20085 | M | 1 | 56 | NR | NR | NR | NR | -Colon (58) | NR | MSH2 (c942. 3A>T) |
| South et al., 20085 | M | 1 | 50 | NR | NR | NR | NR | -Colon (60) | NR | MSH2 (Deleción exones 1-6) |
| Frogatt et al., 199510 | M | 1 | 50 | NR | NR | NR | NR | Colon (50)Útero (50) | NR | Enlace MSH2 |
| Informe actual | V | 1 | 38 | Nariz | MSH2/MSH6 | NP | No | Ninguno | -Adenocarcinoma de colon (padre) | MSH2 (c142 G>T) |
| Informe actual | V | 2 | 38 | Cuello | MSH2/MSH6 | NP | Adenomas sebáceos | Carcinoma urotelial (41) | Ninguno | MSH2 (c.(?_68)_1276+?del) |
IHC: inmunohistoquímica; QA: queratoacantoma; M: mujer; MMR: mismatch repair protein expression (expresión de las proteínas reparadoras de desajustes); NR: no reportado; V: varón.
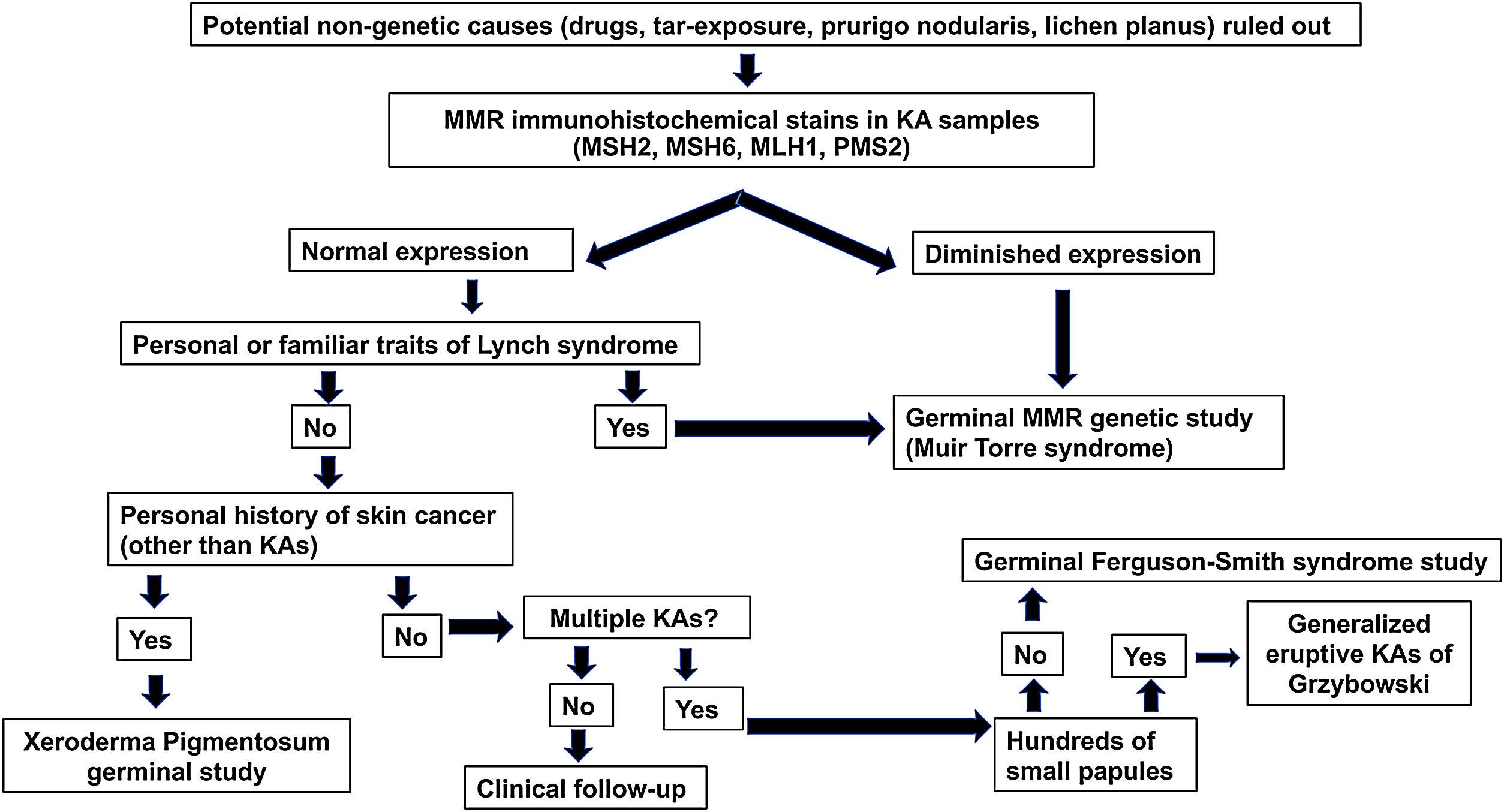
Se propone un algoritmo diagnóstico (fig. 2) para los pacientes de 50 años o menos que desarrollan QA esporádicos. Dicho algoritmo se basa en tres pilares: la presencia de antecedentes familiares de QA/cáncer colorrectal, tinciones inmunohistoquímicas para las proteínas MMR y estudios genéticos de los genes MMR y las alteraciones germinales en el gen TGFBR1.
Los autores declaran no tener ningun conflicto de intereses.
Quisiéramos agradecer a los Doctores Marta Pineda, Joan Brunet, Eleanor Reavey y David Goudie su ayuda prestada con relación al estudio genético de los casos reportados en este trabajo, y sus comentarios para mejorar el mismo.
Por favor, cite este artículo como: Toll A, Sitjas D, Morgado-Carrasco D, Pujol RM. Sporadic Kerathoacantomas in Young Patients: A Case Series and a Proposed Diagnostic Algorithm. Actas Dermosifiliogr. 2022;113:95–98.


