








El xantogranuloma juvenil es un trastorno benigno poco frecuente, que pertenece al amplio grupo de las histiocitosis de células no Langerhans. Se presenta con uno o más nódulos eritematosos o amarillentos, ubicados preferentemente en la cabeza y el cuello. La mayoría de los casos se inician durante el primer año de vida, incluyendo lesiones congénitas. La afectación extracutánea es rara, sugiriéndose tradicionalmente en la literatura estudiar el compromiso ocular. El diagnóstico del xantogranuloma juvenil es fundamentalmente clínico, sin embargo, en ocasiones se requiere confirmarlo con biopsia de piel. Las lesiones cutáneas son autolimitadas, por lo que suelen no requerir tratamiento. En la presente revisión se describen los distintos aspectos clínicos y terapéuticos de esta enfermedad, resaltando la evidencia respecto al estudio diagnóstico del compromiso extracutáneo.
Juvenile xanthogranulomas (JXGs) are rare, benign lesions that belong to the large group of non-Langerhans cell histiocytoses. JXG presents with 1 or more erythematous or yellowish nodules that are usually located on the head or neck. Most JXG lesions are congenital or appear during the first year of life. Extracutaneous involvement is rare, but the literature traditionally suggests investigating the possibility of ocular compromise. JXG is mainly a clinical diagnosis, but a skin biopsy may sometimes be needed for confirmation. JXGs on the skin are self-limiting and usually do not require treatment. This review describes the clinical and therapeutic aspects of JXG, emphasizing available evidence and the diagnosis of extracutaneous involvement.
El xantogranuloma juvenil (XGJ) es una enfermedad benigna poco frecuente y la forma más común de las histiocitosis de células no Langerhans (HCNL)1,2. Pertenece específicamente al grupo C, de las histiocitosis mucocutáneas y surge a partir de una proliferación de células dendríticas, que son positivas para el factor XIIIa, el cual se presume que es el precursor de muchos trastornos dendríticos de HCNL2,3. La primera descripción de la enfermedad fue en 1905, por Adamson, que reportó el caso de un niño de 2 años con múltiples lesiones xantomatosas congénitas4. Siete años más tarde, McDonagh describió cinco nuevos casos muy similares5. Sin embargo, no fue hasta la década de 1950, donde Helwig y Hackney lo denominaron por primera vez XGJ, a través de una serie de 140 casos descritos6.
El XGJ suele manifestarse con lesiones cutáneas tipo pápulas o nódulos eritematosos amarillentos, ubicados principalmente en cabeza y cuello, siendo muy inhabitual el compromiso extracutáneo, el que suele ser a nivel ocular2,7,8.
A continuación, se describen los principales aspectos epidemiológicos, etiopatogénicos, clínicos, diagnósticos y terapéuticos de los XGJ, resaltando la evidencia respecto al estudio del compromiso extracutáneo. Además, se propone un algoritmo de manejo y estudio de compromiso extracutáneo.
EpidemiologíaLa incidencia real del XGJ es desconocida, estimándose una prevalencia de aproximadamente uno por millón de niños, la que se cree puede ser mucho mayor, considerando que las lesiones cutáneas pequeñas y aisladas pueden subdiagnosticarse9. En el Registro de tumores pediátricos de Kiel, que abarcó una cohorte de 35 años, el XGJ representó 129 (0,5%) de las 24.600 lesiones pediátricas10.
El XGJ es una enfermedad principalmente pediátrica, manifestándose solo el 10% en adultos. El 64% por ciento de las lesiones cutáneas se desarrollan antes de la edad de 7 meses y el 85% antes del año. Se ha descrito que hasta un tercio de las lesiones pueden ser congénitas2,9. Se ha observado que la edad de presentación más temprana favorece formas más difusas de la enfermedad. Tiene un predominio en el sexo masculino, con una relación hombres: mujeres de 1,1-7:1, sin observarse diferencias en cuanto a sexo en los XGJ adultos1,10 (tabla 1).
Características epidemiológicas, clínicas e histológicas de los xantogranulomas juveniles
| Epidemiología | -Incidencia real desconocida.-Prevalencia de 1 por millón de niños.-85% de lesiones se desarrollan antes del primer año de vida.-Un tercio de las lesiones pueden ser congénitas.-Relación hombres: mujeres es de 1-7:1. |
| Presentación clínica | -Pápula o nódulo bien definido, de superficie lisa, consistencia firme y coloración roja y amarilla o anaranjada.-Lesión única en 80% de casos.-Ubicación más común en cabeza y cuello.-Formas atípicas: generalizadas, liquenoides, gigantes, mixtas, en placa, subcutáneas, anulares, musculares, agminadas, tipo cuerno cutáneo. |
| Dermatoscopia | -«Patrón en sol poniente»: área central amarillo-anaranjada, con zonas amarillo pálidas y halo eritematoso.-Pueden observarse telangiectasias lineales periféricas. |
| Histología | -Infiltración linfohistiocítica dérmica densa y bien delimitada, con células gigantes (de Touton, cuerpo extraño, en vidrio esmerilado o inespecíficas).-Inmunohistoquímica con tinción positiva para: CD68, CD163, KiM1P, anti-FXIIIa, vimentina y anti-CD4. Tinción negativa para: S-100, CD1a y CD207. |
| Manifestaciones extracutáneas | -El compromiso extracutáneo más frecuente es a nivel ocular.-Compromiso extracutáneo menos frecuente: bazo, pulmones y el sistema nervioso central.-Compromiso extracutánea raro: corazón, médula ósea, tracto gastrointestinal, páncreas, huesos, retroperitoneo, glándulas adrenales, testículos y riñones.-Pueden estar presentes hasta en el 10% de pacientes con neurofibromatosis tipo 1 |
La patogenia del XGJ permanece incierta, sin embargo, clásicamente se ha entendido como un proceso no neoplásico, de tipo reactivo a una injuria inespecífica, como trauma o una infección viral11, lo que contrasta con nueva evidencia que describe ciertos factores genéticos asociados12,13.
El motivo de la lipidización progresiva de los histiocitos, en ausencia de hiperlipidemia, no está claro. En adultos con XGJ se ha demostrado una captación de lipoproteínas de baja densidad y un aumento de síntesis de colesterol dentro de los macrófagos14.
Estudios recientes, en base a secuenciación de exoma completo, sugieren un rol en la activación patológica de la vía de quinasa regulada por señales extracelulares (ERK). La activación de la vía ERK a nivel germinal o en las células madre de la médula ósea, pueden conducir a una enfermedad multiorgánica agresiva, mientras que su activación en una etapa posterior de diferenciación celular, causarían una enfermedad limitada a pocos o solo un órgano y a lesiones unifocales, como sucede en el caso del XGJ12. Chakraborty et al.13 identificaron 17 mutaciones somáticas en 4 lesiones de XGJ, con una mediana de 4 mutaciones por caso, y si bien, no se identificaron mutaciones en BRAF-V600E, como sucede en el caso de las histiocitosis de células de Langerhans (HCL), en un paciente se encontró una mutación de PI3KCD y en otro caso, asociado a neurofibromatosis tipo 1 (NF-1), una mutación germinal del gen que codifica la neurofibromina.
Presentación clínica- a.
Manifestaciones cutáneas:
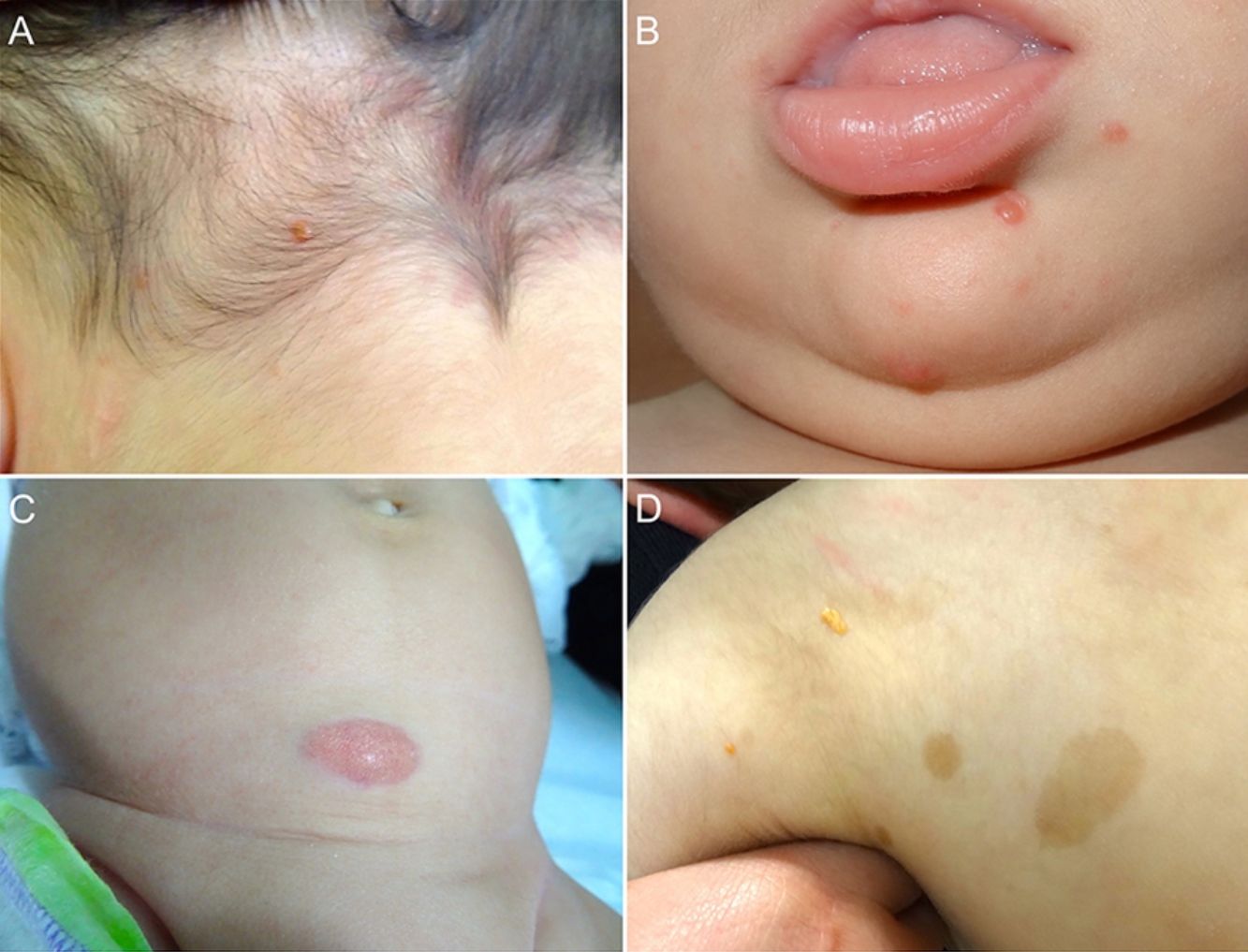
Las formas cutáneas son la presentación más frecuente del XGJ. Se han descrito dos clasificaciones según el tamaño de las lesiones: 1) Caputo et al.15 distinguieron tres tipos principales: nodular pequeño o papular (2-5mm), nodular grande (5-20mm) y xantogranuloma gigante (> 20mm); 2) Gianotti et al.16 agruparon los XGJ en dos formas de presentación: micronodular (<10mm de tamaño, usualmente múltiples lesiones) y macronodular (10-20mm) (tabla 2). Si bien clínicamente pueden presentar un espectro amplio de lesiones, en cuanto a forma, coloración y número, hasta en un 80% de los casos se presenta como una lesión cutánea única. Los XGJ múltiples son más frecuentes en niños y van desde 2 o 3 lesiones a más de 1003,17. La lesión típica consiste en una pápula o nódulo bien definido, de superficie lisa, con coloración rosa-rojiza al inicio, que luego adquiere la coloración clásica amarilla, naranja o marronácea; tienen consistencia firme y algunos presentan telangiectasias superficiales, siendo generalmente asintomáticos, con excepción de algunas localizaciones especiales o cuando son muy grandes. La ubicación anatómica más común es la cabeza y el cuello, seguido del tronco y las extremidades1,18,19 (fig. 1A, B) (tabla 1).
 Lactante masculino de un año con XGJ único en región occipital. B) Lactante masculino de 8 meses con XGJ múltiples en mentón, congénitos. C) Lactante femenina de 4 meses con un XGJ congénito gigante en abdomen. D) XGJ asociado a manchas café con leche, en un preescolar de 4 años con neurofibromatosis tipo 1.")
A) Lactante masculino de un año con XGJ único en región occipital. B) Lactante masculino de 8 meses con XGJ múltiples en mentón, congénitos. C) Lactante femenina de 4 meses con un XGJ congénito gigante en abdomen. D) XGJ asociado a manchas café con leche, en un preescolar de 4 años con neurofibromatosis tipo 1.
Las variantes atípicas de XGJ incluyen formas generalizadas, liquenoides, gigantes (>2cm de diámetro), mixtas, en placa, subcutáneas, anulares, musculares, agrupadas y casos raros tipo cuerno cutáneo20,21.
Con respecto a los XGJ congénitos, la presentación clínica parece ser aún más variada que la de los XGJ clásicos. Suelen ser tumores grandes y solitarios, de morfología atípica, describiéndose en la literatura casos de lesiones subcutáneas profundas, placas infiltrativas, tumores exofíticos y placas agminadas, entre otros22 (fig. 1C). Las dos complicaciones más comunes del XGJ congénito son la cicatrización atrófica y la ulceración, esta última, aparentemente con un mayor riesgo en lesiones exofíticas grandes22,23.
- b.
Manifestaciones extracutáneas:
Aunque las lesiones de XGJ se encuentran limitadas a la piel en la mayoría de los pacientes, se han descrito múltiples casos de afectación extracutánea aislada o de varios órganos, siendo más común el compromiso ocular. Un estudio retrospectivo reciente, que analizó las distintas series y casos publicados de XGJ en la literatura, encontró que, del total de 2949 casos pediátricos, solo el 0,24% tuvo compromiso intraocular. Por esto, concluyen que el rendimiento de derivar a un niño con XGJ cutáneo sin ningún síntoma o signo ocular o visual para una evaluación oftalmológica parece ser extremadamente bajo24. Otros autores describieron que aproximadamente el 40% de los pacientes con XGJ ocular tenían lesiones cutáneas previas. Estas pueden aparecer antes o después del diagnóstico oftalmológico, con un retraso de 8 a 10 meses25.
El XGJ ocular es casi siempre unilateral y afecta más comúnmente al iris, aunque también se ha descrito afectación de la órbita, el nervio óptico, el coroides y la conjuntiva. El síntoma de presentación más frecuente es el ojo rojo unilateral, seguido por un tumor en el iris o conjuntival. A diferencia de las lesiones cutáneas, el compromiso intraocular no se resuelve espontáneamente y puede provocar hifema, glaucoma y/o pérdida visual17,25,26. Tres factores de riesgo se han sugerido para la afectación ocular: 1) la forma micronodular cutánea de XGJ, 2) lesiones cutáneas múltiples y 3) edad temprana de presentación (≤2 años)16,17. En base a esto, sugerimos derivar a oftalmología, en caso de 2 o más XGJ cutáneos o en lesiones de inicio temprano.
El XGJ sistémico es un trastorno histiocítico raro, que se presenta usualmente con múltiples nódulos cutáneos y/o subcutáneos, asociado al compromiso de dos o más órganos viscerales27. A la fecha, solo se han descrito 61 casos de XGJ sistémico, donde los órganos más frecuentemente afectados han sido: el hígado, el bazo, los pulmones, los ojos y el sistema nervioso central, con casos anecdóticos a nivel del corazón, la médula ósea, el tracto gastrointestinal, el páncreas, los huesos, el retroperitoneo, las glándulas adrenales, los testículos y los riñones1,28–30,31. La edad de presentación es más temprana que el XGJ clásico, con un promedio 5,5 meses y una relación H:M de 1,4:1. Se ha propuesto que las lesiones cutáneas múltiples son un factor de riesgo para el compromiso sistémico, encontrándose este hallazgo en el 67% de los pacientes con XGJ sistémico, teniendo un período de latencia de meses a varios años24,29. Por lo tanto, un estudio de compromiso extracutáneo inicialmente negativo en pacientes con múltiples XGJ, no excluye el desarrollo de enfermedad sistémica en un momento posterior, siendo razonable mantener el estudio diagnóstico en el tiempo.
En el momento del diagnóstico, se debe realizar un examen físico meticuloso para determinar la existencia de compromiso sistémico32. Los signos y síntomas dependen del órgano afectado y van desde un curso asintomático a uno letal. Particularmente, el compromiso del sistema nervioso central y la enfermedad hepática se han asociado con una morbimortalidad significativa28,29.
Los XGJ ocurren en aproximadamente el 5-10% de los pacientes que padecen NF-133 (fig. 1D). Esto se encuentra condicionado a la edad del niño, dado que los XGJ tienden a involucionar a la edad en que la NF-1 se vuelve clínicamente evidente. Si bien la relación entre XGJ y NF-1 aún no está del todo clara, hay varios autores que promueven y enfatizan la búsqueda de estigmas de NF-1 en niños con XGJ múltiples34. Se ha reportado una asociación triple de XGJ, NF-1 y leucemia mielomonocítica juvenil (LMMJ), lo que es objeto de frecuentes debates. En 1995, Zvulunov et al.35 concluyeron que esta triple asociación es 30-40 veces más común de lo esperado por el azar y estimó un riesgo de desarrollar LMMJ 20 a 32 veces mayor en niños con NF-1 y XGJ versus aquellos con solo NF-1. No obstante, las limitaciones metodológicas y estadísticas de este estudio podrían haber resultado en una sobreestimación de la asociación. En otras dos series retrospectivas posteriores, no se encontró una mayor incidencia de LMMJ en pacientes con XGJ y NF-136,37, siendo ratificado por un estudio retrospectivo reciente de casos y controles, donde se evaluaron 739 pacientes con NF-1 en un período de 20 años, reportando una prevalencia de neoplasias malignas de 1,9% (14/739), encontrando XGJ en 28,5% (4/14) de los casos versus un 21% (6/29) de los controles, sin diferencia estadísticamente significativa33. La ausencia de una relación entre malignidad y XGJ en niños con NF-1 puede brindar tranquilidad a los médicos y a los padres. Debido a que tanto el XGJ como las neoplasias malignas hematológicas y sólidas están asociados con NF-1, su coexistencia en el mismo paciente podría ser solo una coincidencia33,38. Los pacientes con NF-1 deben ser monitorizados por malignidad independiente de la presencia o ausencia de XGJ, sobre todo tumores malignos de la vaina nerviosa periférica39.
Diagnóstico- a.
Clínico:
El diagnóstico de XGJ es fundamentalmente clínico, de acuerdo a la historia, evolución y morfología de las lesiones. En algunos casos se requiere estudio histopatológico e inmunohistoquímico para confirmar el diagnóstico2,3.
- b.
Dermatoscopia:
La dermatoscopia resulta ser una técnica no invasiva útil en el diagnóstico clínico de XGJ. Se ha descrito un patrón denominado «sol poniente», que se caracteriza por un área central amarillo-naranja, que puede mostrar áreas con «nubes» de depósitos de color amarillo pálido y un halo eritematoso, lo que se puede observar en cualquier etapa de la evolución40 (fig. 2). También se han descrito telangiectasias lineales periféricas y otras características no específicas, que incluyen: red discreta de pigmento, líneas blanquecinas y vasos finos y ramificados40,41.
- c.
Histología:
.")
La histología clásica del XGJ muestra una infiltración celular densa, en forma de lámina, no encapsulada y bien delimitada, en la dermis y la porción superior del tejido celular subcutáneo, mientras que la epidermis y las estructuras cutáneas anexas están indemnes. El infiltrado celular incluye cinco tipos de células principales (vacuoladas, xantomatosas, en forma de huso, festoneadas y oncocíticas) en proporciones variables, de variantes monomorfas a mixtas, con diferentes tipos de células gigantes (inespecíficas, cuerpo extraño, Touton y «vidrio esmerilado»)11 (fig. 3A y B). La apariencia microscópica depende principalmente del tiempo de evolución de la lesión: mientras que las lesiones tempranas muestran un infiltrado monomórfico de macrófagos libres de lípidos que pueden ocupar la mayor parte de la dermis, las lesiones maduras contienen abundantes macrófagos espumosos vacuolados y células gigantes multinucleadas de tipo Touton, particularmente en la superficie dermis1,11.
 Infiltrado dérmico de histiocitos y eosinófilos (H/E 40x y 100x, respectivamente). C) Inmunohistoquímica: marcador CD68 positivo.")
Las células de Touton son típicas de XGJ y se caracterizan por tener un anillo de núcleos alrededor del citoplasma de alto contenido de lípidos. Sin embargo, no se observan en todos los casos, pudiendo estar ausentes hasta en un 15% de los pacientes. Clásicamente se describen las células de Touton como patognomónicas de XGJ, pero pueden encontrarse también en otras lesiones xantomatosas e histiocíticas (enfermedad de Erdheim Chester y xantogranuloma necrobiótico), e incluso dermatofibromas1,2.
Respecto a la inmunohistoquímica, las lesiones de XGJ se tiñen con marcadores de macrófagos, que incluyen: CD68, CD163, KiM1P, anti-FXIIIa, vimentina y anti-CD4, y generalmente son negativos para S-100, CD1a y CD207 (antilangerina), que es específico para las células de Langerhans11,42 (fig. 3C).
La microscopia electrónica muestra histiocitos suaves con muchas gotas de lípidos intracitoplasmáticos. Se han descrito otros hallazgos intracitoplasmáticos no específicos, como cuerpos densos, parecidos a gusanos y palomitas de maíz. No se observan los gránulos de Birbeck característicos de las HCL43.
Se ha sugerido como una técnica diagnóstica útil en XGJ, la citología por aspiración con aguja fina. Los frotis citológicos muestran histiocitos mononucleados lipidizados finamente vacuolados y multinucleados con núcleos reniformes u ovales en un fondo de infiltrado inflamatorio de células mixtas, con una proporción variable de linfocitos, neutrófilos y eosinófilos, junto con células gigantes de tipo Touton o cuerpo extraño. Sin embargo, se requiere un alto índice de sospecha por parte del citopatólogo44.
- d.
Ecografía doppler:
Las técnicas no invasivas, como la ecografía doppler, pueden ser útiles en la evaluación y monitorización de las lesiones, como una alternativa a la histología. En el examen ultrasonográfico, se describe un nódulo hipoecoico, bien delimitado, ubicado en el límite dermoepidérmico o dermis, sin realce posteror ni sombra lateral y en algunos casos, con doppler color que muestra vascularización dentro de la lesión, con vasos arteriales finos y de baja velocidad (fig. 4)45,46.
Estudio del compromiso extracutáneo
Considerando que la afectación extracutánea es rara, no existe un consenso sobre la pertinencia de realizar pruebas complementarias en ausencia de signos o síntomas sugestivos32. Höck et al.11 sugieren que, dependiendo de la historia, los estudios diagnósticos pueden incluir un análisis de laboratorio, una ecografía de ganglios linfáticos y de abdomen, radiografía de tórax, examen radiográfico esquelético o una exploración ósea con radionúclidos, tomografía computarizada o una resonancia magnética del cerebro y un examen oftalmológico, además del examen completo de la piel. Por otro lado, basado en el caso de un lactante de 2 meses con afectación cutánea, pulmonar y hepática, Meyer et al.47 sugieren que todos los niños con ≥ 2 XGJ cutáneos deben someterse a una evaluación de XGJ sistémico, incluyendo tomografía computarizada o resonancia magnética de cerebro/tórax/abdomen. Si se sospecha una lesión cardíaca, se debe realizar una ecocardiografía doppler. La evaluación de laboratorio debe incluir un hemograma completo (que puede conducir a una aspiración/biopsia de médula ósea si hay citopenias), un perfil metabólico completo que incluya pruebas de función renal y hepática.
Considerando los escasos reportes de XGJ sistémico y la evolución de carácter benigno y autolimitada de las lesiones cutáneas y extracutáneas, nos parece coherente realizar el estudio diagnóstico, según la sintomatología, el examen físico y los órganos más comúnmente afectados (fig. 5). Sin embargo, se requiere el diseño de estudios que permitan mejorar la evidencia para entregar recomendaciones más precisas.
Diagnóstico diferencial
El diagnóstico diferencial del XGJ depende de su presentación clínica y sitio afectado e incluye enfermedades benignas y malignas.
Una de las principales entidades con las que se debe diferenciar, considerando su tratamiento y pronóstico, son las HCL, sobre todo en casos de lesiones cutáneas múltiples y XGJ sistémico. La histología permite el diagnóstico, dado que en la HCL se observan núcleos celulares en forma de «grano de café», además de la inmunohistoquímica con marcadores S-100, CD1a y CD207 positivos. Existen varios reportes de XGJ en pacientes con HCL, con un período de diferencia menor a 5 años, lo que estaría explicado por una relación biológica entre ambas entidades o un mecanismo desconocido de transformación de la HCL a XGJ, mediado por la quimioterapia, dado que la mayoría de los reportes habían sido tratados o se encontraban en tratamiento con quimioterapéuticos cuando desarrollaron el XGJ48,49.
Otro diagnóstico diferencial importante son algunas formas de HCNL como la histiocitiosis cefálica benigna, el xantogranuloma necrobiótico y la reticulohistiocitosis multicéntrica2,27,50. El XGJ de tipo nodular pequeño puede ser difícil de distinguir de la histiocitosis cefálica benigna, sin embargo, se han sugerido algunas características clínicas a favor de esta última, tales como: la distribución simétrica de sus lesiones, la progresión cefalocaudal, el infrecuente compromiso palmo-plantar, nasal y de cuero cabelludo, además del nulo compromiso sistémico51.
También pueden simular ser XGJ lesiones tales como: neurotecoma celular pediátrico, xantomas tuberosos, moluscos contagiosos, mastocitosis cutánea máculo-papular, mastocitomas, hemangiomas, neurofibromas, dermatofibromas, nevos de spitz, histiocitomas, reticulohistiocitosis congénita, sarcoidosis, xantomas, entre otros9,52–54.
TratamientoCon respecto a la terapia, se debe considerar que los XGJ normalmente siguen un curso benigno, con remisión espontánea en aproximadamente 3-6 años, por lo que no suelen requerir tratamiento1,2. Sin embargo, en algunos casos de xantogranulomas de inicio en la adultez, las lesiones tienden a persistir, con menos probabilidad de autoinvolución55.
En caso de deterioro funcional o alta tensión psicológica debido al compromiso visible, sobre todo en los padres, las lesiones solitarias pueden extirparse quirúrgicamente o tratarse con otros métodos destructivos, como el láser de CO2. Para el caso de las lesiones múltiples y asintomáticas, la política de «esperar y ver» es la primera opción en la gran mayoría de los casos11.
En los casos de XGJ ocular, se suelen usar corticoides tópicos o intralesionales y en caso de lesiones rápidamente progresivas o el desarrollo de complicaciones, como el glaucoma o el hifema, se puede requerir el uso de corticoides sistémicos o cirugía, existiendo algunos escasos reportes con buena respuesta a radioterapia y quimioterapia25,29.
En XGJ sistémico, las indicaciones de tratamiento dependen del grado de disfunción visceral que generen estas lesiones benignas. La estrategia de «esperar y ver» se recomienda en la mayoría de los casos, considerando que muchos de estos tendrán un curso autolimitado. De esta forma, la terapia debe iniciarse cuando el XGJ interfiere con las funciones vitales de los órganos. Se informan diversas estrategias de tratamiento que incluyen: resección quirúrgica, radioterapia y/o quimioterapia con esquemas usados en HCL, con prednisona y vinblastina, y en casos refractarios citarabina y 2-clorodeoxiadenosina3,29,53. Sin embargo, debido al número limitado de casos reportados, no se cuenta con pautas de tratamiento, requiriéndose un tratamiento individualizado y multidisciplinario.
ConclusiónEL XGJ es una entidad poco frecuente, con un amplio espectro de manifestaciones clínicas, que requiere una evaluación exhaustiva que permita el diagnóstico del compromiso extracutáneo. Para ello, de acuerdo con la poca evidencia disponible, el estudio diagnóstico dirigido según los hallazgos clínicos, parece ser una estrategia razonable en el manejo de estos pacientes. El tratamiento suele ser expectante, sin embargo, en casos sistémicos se puede requerir el uso de agentes quimioterapéuticos, siendo necesario el diseño de estudios que evalúen los distintos esquemas, para optimizar su uso específico en esta multifacética enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


