









Proteins belonging to the RAS/mitogen activated protein kinase (MAPK) pathway play key roles in cell proliferation, differentiation, survival, and death. For more than 30years now we have known that 30% of human cancers carry somatic mutations in genes encoding proteins from this pathway. Whereas somatic mutations have a high malignant potential, germline mutations are linked to developmental abnormalities that are often poorly clinically differentiated, although each is dependent upon the specific gene affected. Thus, all patients share varying degrees of mental retardation or learning difficulties, heart disease, facial dysmorphism, skin anomalies, and, in some cases, predisposition to cancer. These syndromes, known as rasopathies, include Noonan syndrome, Costello syndrome, neurofibromatosis-1, LEOPARD syndrome, cardiofaciocutaneous syndrome, and Legius syndrome. Recognizing the skin manifestations of rasopathies can facilitate diagnosis of these syndromes.
Las proteínas de la vía RAS/MAPK (mitogen activated protein kinase pathway) desempeñan un papel fundamental en la proliferación, diferenciación, supervivencia y muerte celular. Desde hace más de 30 años se sabe que el 30% de los cánceres humanos presentan una mutación somática en alguno de los genes que codifican estas proteínas. En contraste con el elevado potencial de malignidad de las mutaciones somáticas, las mutaciones en la línea germinal provocan anomalías en el desarrollo del individuo que, si bien dependen específicamente del gen afectado, a menudo se superponen clínicamente. Así, todos los pacientes comparten un grado variable de retraso mental o dificultades de aprendizaje, trastornos cardiacos, dismorfismo facial, anomalías cutáneas y, en algunas instancias, predisposición al cáncer. Entre estos síndromes, conocidos como rasopatías, se incluyen el síndrome de Noonan, el síndrome de Costello, la neurofibromatosis 1, el síndrome LEOPARD, el síndrome cardio-facio-cutáneo y el síndrome de Legius. Es interesante conocer las manifestaciones cutáneas de las rasopatías, ya que estas pueden ayudar a esclarecer el diagnóstico de la enfermedad.
RAS genes play an essential role in signaling through the mitogen-activated protein kinase (MAPK) pathway, which regulates cell proliferation, differentiation, survival, and death. These genes were first identified in tissue samples from bladder and lung cancer and were so-named because of their sequence homology with the Harvey (HRAS) and Kirsten (KRAS) rat sarcoma viruses (RAS being an abbreviation of rat sarcoma).1 Each of the genes encoding RAS/MAPK proteins is located on a different chromosome and encodes a different protein, and consequently mutations in each will lead to different diseases2 (Table 1).
Molecular Characterization of the RASopathies.
| Syndrome | Gene | Chromosome | Protein | Protein Function |
| Noonan | PTPN11 | 12q24.1 | SHP2 | Phosphatase |
| SOS1 | 2p22.1 | SOS1 | RasGEF | |
| KRAS | 12p12.1 | KRAS | GTPase | |
| RAF1 | 3p25.1 | CRAF | Kinase | |
| MAP2K1 | 15q22.31 | MEK1 | Kinase | |
| Cardiofaciocutaneous | KRAS | 12p12.1 | KRAS | GTPase |
| BRAF | 7q34 | BRAF | Kinase | |
| MAP2K1 | 15q22.31 | MEK1 | Kinase | |
| MAP2K2 | 19p13.3 | MEK2 | Kinase | |
| Costello | HRAS | 11p15.5 | HRAS | GTPase |
| KRAS | 12p12.1 | KRAS | GTPase | |
| BRAF | 7q34 | BRAF | Kinase | |
| MAP2K1 | 12p12.1 | MEK1 | Kinase | |
| Type-1 neurofibromatosis 1 | NF1 | 17q11.2 | Neurofibromin | RasGAP |
| Legius | SPRED1 | 15q14 | SPRED1 | Sprouty-related EVH1 domain-containing protein1 |
| LEOPARD | PTPN11 | 12q24.1 | SHP2 | Phosphatase |
| RAF1 | 3p25.1 | CRAF | Kinase | |
| Capillary malformation-arteriovenous malformation | RASA1 | 5q14.3 | P120Gap | RasGAP |
| Autoimmune lymphoproliferative | NRAS | 1p15.2 | NRAS | GTPase |
| Gingival fibromatosis 1 | SOS1 | 2p22.1 | SOS1 | RasGEF |
Modified from Tydiman et al.2
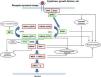
The RAS protein family is a subfamily of the small G proteins and contains specific members, such as the classical RAS proteins HRAS, KRAS, and NRAS, and others such as R-RAS, TC21, M-RAS, Rap IA, Rap IB, Rap2A, Rap 2B, RaIA, and RaIB.3 RAS proteins function as essential mediators in the transduction of extracellular signals, and their activation state is determined by binding to guanosine triphosphate (GTP) or guanosine diphosphate (GDP), which generates the active and inactive forms, respectively.4,5 The stimulation of cell receptors via cytokines, calcium channels, integrins, heteromeric G-protein receptors, growth factors, and other peptides leads to dissociation of GDP from the RAS protein and binding of GTP, leading to activation of RAS and the promotion of interactions with various effector proteins such as RAF and MEK.6,7 GDP/GTP exchange is stimulated by guanine nucleotide exchange factors (GEF) such as son-of-sevenless (SOS), RAS guanine release factor (RASGRF), and RAS guanyl releasing protein (RASGRP); when the receptor is stimulated, it binds the Src homology 2 (SH2) domains of SHC, SHP2, and GRB2, which recruit cytoplasmic SOS, and this in turn promotes exchange of GDP/GTP in RAS proteins. RAS-protein activation is limited by GTPase activity, which is both intrinsic to RAS and stimulated by GTPase activator proteins (GAPs) that regulate the exchange between the active form bound to GTP and the inactive form bound to GDP (Fig. 1). Activation of RAS is accompanied by activation of RAF (ARAF, BRAF, and CRAF), which is the first MAPK protein in the pathway; next, it activates ERK1/ERK2, which are the final effectors of the pathway and act on a large number of cytosolic and nuclear proteins that are ultimately responsible for maintaining the cell cycle.8
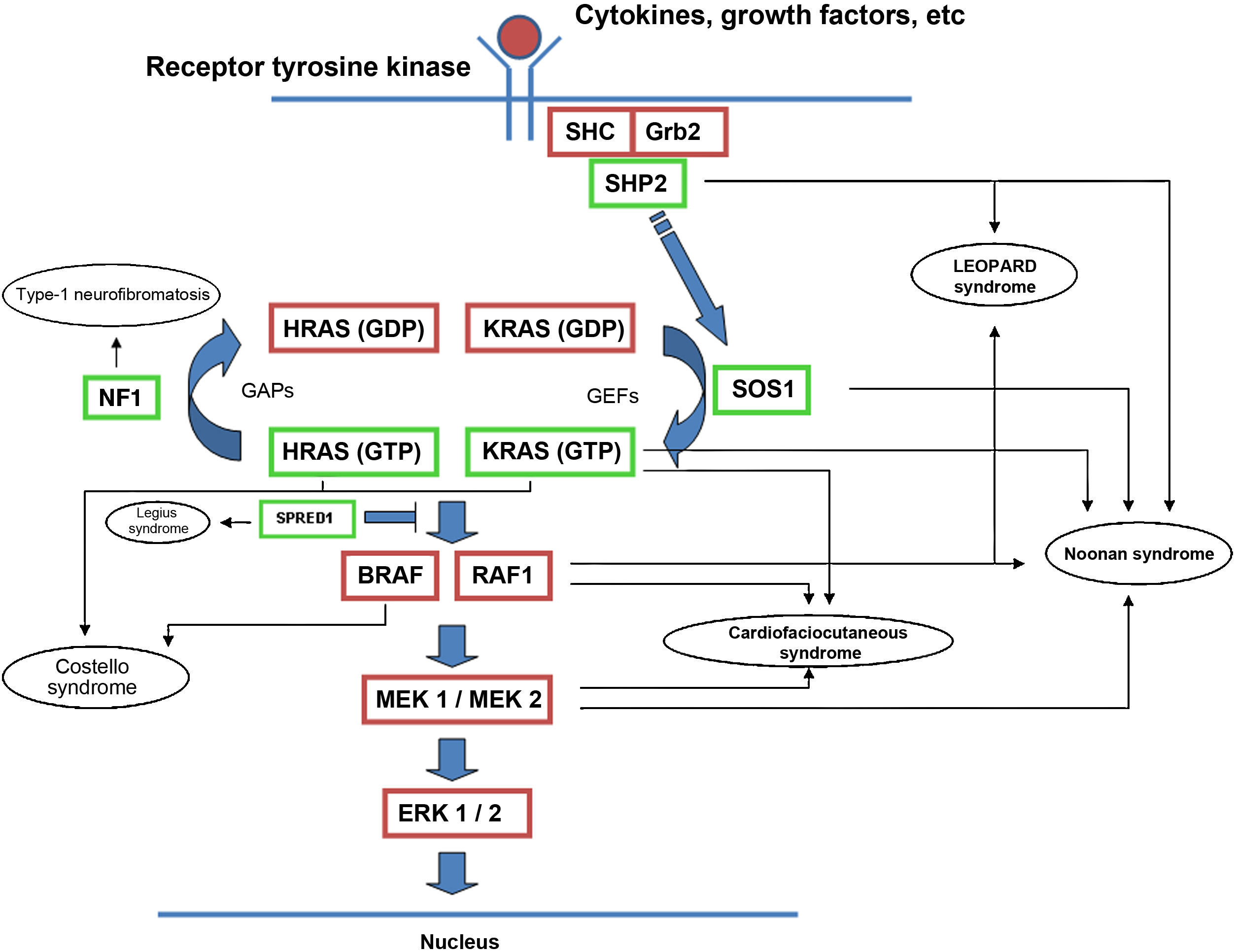
RAS signaling pathway and genetic syndromes associated with abnormalities in the pathway. Following stimulation of cell-surface receptors, intracellular proteins such as SHC, GRB2, and SHP2 are activated and recruit cytoplasmic SOS1. SOS promotes exchange of GTP and GDP in RAS proteins, which are activated by phosphorylation. GTP-bound RAS promotes interaction with other effectors such as RAF and MEK. GDP/GTP exchange is stimulated by guanine nucleotide exchange factors (GEF) and limited by GTPase activating proteins (GAPs) that catalyze the conversion to the inactive GDP-bound form. Activation of RAS proteins is accompanied by activation of RAF (BRAF, RAF1), MEK1A1/MEK1A2, and, ultimately, ERK1/ERK2, which are the final effectors in the RAS/MAPK pathway and responsible for maintenance of the cell cycle.
The RAS/MAPK pathway was initially studied in relation to oncogenesis, since it is dysregulated in 20% to 30% of somatic tumors.9 Unlike somatic mutations in the RAS pathway, which have a very high malignant potential, germline mutations lead to developmental abnormalities that are often poorly clinically differentiated. All patients have some degree of mental retardation or learning difficulties, cardiovascular disease (mainly pulmonary stenosis and myocardial hypertrophy), facial dysmorphism, macrocephaly, short stature, skin abnormalities, and, in some cases, predisposition to cancer (Table 2). Clinical overlap is probably due to the fact that each mutation will have an impact on other components of the RAS/MAPK pathway—with the exception of LEOPARD syndrome, all of the mutations identified to date are characterized by an increase in physiological activity of the mutated protein and therefore increased signaling through the pathway.10
Characteristic Skin Manifestations of the RASopathies.
| Syndromes | Skin Manifestations |
| Noonan | Café-au-lait spots |
| Melanocytic nevus | |
| Lymphedema of the lower limbs | |
| Cardiofaciocutaneous | Short, thin, curly hair |
| Ichthyosiform scaling | |
| Follicular keratosis | |
| Ulerythema ophryogenes | |
| Acquired multiple nevi | |
| Café-au-lait spots | |
| Costello | Loose skin |
| Hyperpigmentation | |
| Papillomatous lesions around the orifices | |
| Deep lines on the palms | |
| Type-1 neurofibromatosis | Café-au-lait spots |
| Axillary and inguinal freckles | |
| Neurofibromas | |
| Xanthogranulomas | |
| Glomus tumors | |
| Pale macules | |
| Diffuse hyperpigmentation | |
| Pruritus | |
| Legius | Café-au-lait spots |
| Freckles | |
| Lipomas | |
| Leopard | Freckles |
| Dark café-au-lait spots | |
| Capillary malformation-arteriovenous malformation | Capillary and arteriovenous malformations |
| Arteriovenous fistulas | |
| Type-1 gingival fibromatosis | Hereditary gingival fibromatosis |
Although the genotype-phenotype relationship is unclear, some authors have proposed the classification of these neuro-cardio-facial-cutaneous syndromes into 3 groups according to the point in the pathway that is affected.11 One group consists of syndromes caused by mutation of upstream genes, that is, PTPN11, SOS1, and NF1. In this group, the patients tend to have a Noonan-type phenotype, mild mental retardation, and a greater likelihood of pigmented lesions than ectodermal manifestations. In addition, in the presence of NF1 and PTPN11 mutations, there appears to be a slightly increased risk of leukemia. The second group comprises syndromes caused by mutations in KRAS or downstream genes. These syndromes mainly affect cognitive function and the skin, which has redundant folds, keratinization defects, and hair abnormalities. Although the risk of associated malignancy is low, leukemia may develop. Finally, there are the syndromes caused by mutations in HRAS, of which the main example is Costello syndrome. These patients commonly have atrial fibrillation, skin hyperpigmentation, papillomatous skin growths, and an increased likelihood of developing soft-tissue tumors.
In addition, there are other diseases associated with germline mutations in the RAS/MAPK pathway. Examples are autoimmune lymphoproliferative syndrome,12 capillary malformation-arteriovenous malformation syndrome,13 and hereditary gingival fibromatosis type 1.14 These syndromes are not associated with general developmental defects, instead affecting immunological mechanisms and the formation of the blood vessels. The identification of these entities further highlights the importance of the RAS/MAPK pathway in human biology.
Skin Manifestations of RASopathiesSpecific skin manifestations cannot be ascribed to individual RASopathies. The most common dermatological findings for each RASopathy (Table 3) can by divided into pigmented lesions (café-au-lait spots, lentigines, and melanocytic lesions), ectodermal lesions (ichthyosiform manifestations, follicular hyperkeratosis, and short, curly, thin hair), and hyperplasia (redundant skin and papillomatous growths).
Malignant Disease Described in Patients With RASopathies.
| Noonan | LEOPARD | Cardiofaciocutaneous | Costello | Type-1 Neurofibromatosis | Legius | |
| JMML | + | |||||
| JMML-like disease | + | + | ||||
| ALL | + | + | + | |||
| AML | + | + | + | |||
| CMML | + | |||||
| CLL | + | |||||
| Lymphoma | + | |||||
| Hepatoblastoma | + | + | ||||
| Rhabdomyosarcoma | + | + | + | |||
| Neuroblastoma | + | + | + | + | + | |
| Wilms tumor | + | + | ||||
| Brain tumor | + | + | ||||
| Testicular tumor | + | |||||
| Bladder carcinoma | + | + | ||||
| Breast cancer | + | + | + | |||
| Pheochromocytoma | + | |||||
| Gliomas | + | |||||
| Neural tumors | + | + | ||||
| Gastrointestinal carcinoma | + | + | ||||
| Lung carcinoma | + | + | ||||
| Ovarian cancer | + | |||||
| Melanoma | + | + |
Modified from Hasle.33 Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphoid leukemia; CMML, chronic myelomonocytic leukemia; JMML, juvenile myelomonocytic leukemia.
Since the role of the RAS/MAPK pathway in the development of the skin remains unclear, the mechanisms underlying these skin abnormalities are not known. However, a recent experimental study has shown that hyperactivation of KRAS blocks proliferation of the hair and induces the appearance of redundant skin, papillomatous growths, and short nails.15
Noonan SyndromeNoonan syndrome (Online Mendelian Inheritance in Man [OMIM] code 163950) is a genetically heterogeneous autosomal recessive disorder. To date, mutations have been described in PTPN11, SOS1, KRAS, RAF1, and BRAF,16,17 but mutations in other genes are very probably also involved. Approximately half of all cases have a mutation in PTPN11, a gene with 16 exons on chromosome 12q24.1.18PTPN11 encodes the protein tyrosine phosphatase SHP2, whose catalytic activity is autoinhibited through an interaction between 2 of its own domains, the N-terminal SH2 domain and the catalytic protein tyrosine phosphatase domain. This interaction is affected by most mutations in PTPN11, and as a result control of the catalytic activity of the protein is lost, leading to overactivity of the RAS/MAPK pathway.19 A substantial number of patients with LEOPARD syndrome also have mutations in PTPN11,20 which accounts for its similarity with Noonan syndrome. The second most common cause of Noonan syndrome (in approximately 17% to 28% of cases) is a mutation in SOS1.21,22SOS1 encodes a protein with GEF activity that stimulates the conversion of inactive, GDP-bound RAS to the active, GTP-bound form. The mutations increase the activity of SOS1 protein and therefore overstimulate the RAS/MAPK pathway. The same effect occurs with the less-frequently observed mutations in KRAS and RAF123 and BRAF.17 Finally, cases of Noonan syndrome have recently been described in patients with mutations in NRAS.24 Patients with a phenotype similar to Noonan syndrome have also been reported to carry mutations in SHOC2, which encodes a protein involved in binding of RAS to downstream effectors,25 and in CBL,26 a tumor-suppressor gene that is mutated in myeloid leukemias.
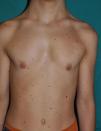
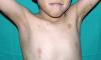
The essential phenotypic characteristics of Noonan syndrome include facial abnormalities, short stature, congenital heart defects, coagulopathies, and varying degrees of mental retardation. Facial characteristics include widened forehead, ocular hypertelorism, ptosis, epicanthus, high philtrum, and high wide peaks of the vermillion border of the upper lip (Fig. 2), low-set, posteriorly rotated ears, arched eyebrows, clear blue eyes, wide neck, and low hairline at the nape of the neck. Pectus excavatum or carinatum is common and the nipples are often widely separated. In 50% to 80% of cases, there are congenital heart defects—this mainly involves pulmonary stenosis (20% to 50%) and hypertrophic cardiomyopathy (20% to 30%), but atrial septal and ventricular defects and tetralogy of Fallot are also seen. Cryptorchidism and hemorrhagic diathesis are observed less frequently. Mental retardation is variable (15% to 35% of patients) and tends not to be severe.27,28 Although there is no strict genotype-phenotype relationship, mutations in PTPN11 are commonly associated with the presence of pulmonary valve stenosis, short stature, hemorrhagic diathesis, and chest deformities, whereas patients who do not carry this mutation tend to have heart abnormalities and less striking facial characteristics.29 Patients with mutations in SOS1 have pulmonary stenosis and marked ectodermal abnormalities, whereas atrial septal defects are not as common as in patients with PTPN11 mutations.21,22 The phenotype associated with mutations in KRAS is less clearly defined and often overlaps with that of Costello syndrome and cardiofaciocutaneous syndrome. Finally, mutations in RAF1 are strongly associated with the presence of hypertrophic cardiomyopathy (found in 76% of patients with RAF1 mutations and in only 18% of other cases of Noonan syndrome) and pigmented skin lesions.23 Mutations in BRAF, on the other hand, appear to be associated with a more severe phenotype and with the presence of dark lentigines and multiple melanocytic nevi.17 In summary, Noonan syndrome is both clinically and genetically heterogeneous and much larger patient series will be required to improve our understanding of the genotype-phenotype relationship.
Skin Manifestations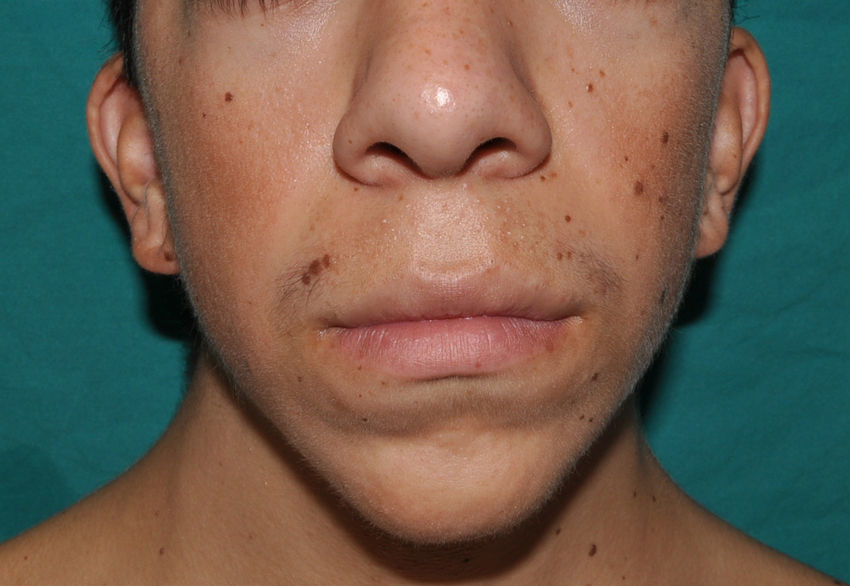
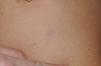
The skin abnormalities observed in patients with Noonan syndrome appear to depend on the mutation responsible. Thus, ectodermal abnormalities (short, curly hair, eyebrow alopecia, and erythema) are much more common in patients with mutations in SOS1,21,22 whereas mutations in SHOC2 are characteristically associated with loose anagen hair.24 Pigmented lesions are more common in patients with mutations in RAF123 or BRAF17 (Fig. 3). Other skin manifestations of Noonan syndrome are lymphatic abnormalities,30 granular cell tumors,31 and capillary malformations.32
Noonan Syndrome and Risk of Cancer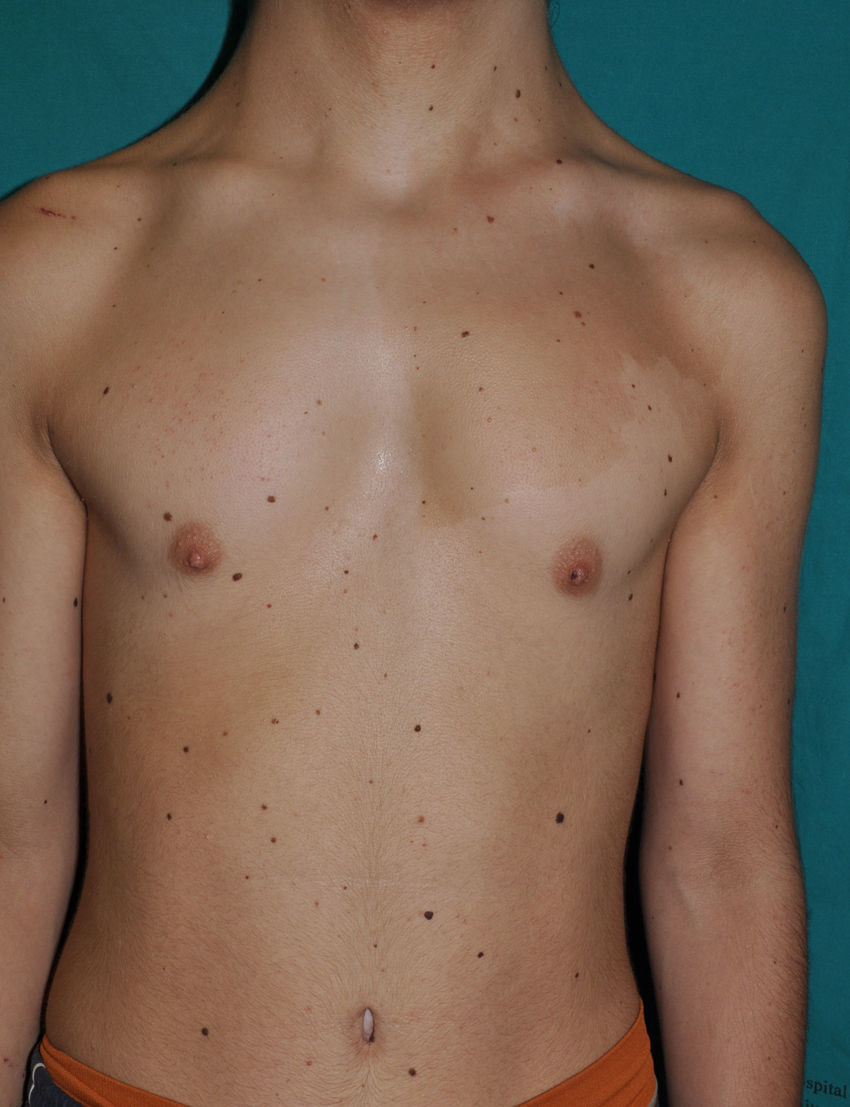
Patients with Noonan syndrome have an increased risk of hematological and other tumors such as rhabdomyosarcoma, neuroblastoma, giant cell tumors, and testicular tumors, although the absolute risk of developing one of these tumors is relatively low.33 Newborns with mutations in PTPN11 can suffer a myeloproliferative syndrome very similar to juvenile myelomonocytic leukemia that remits spontaneously.33 Unlike PTPN11, which acts as an oncogene in certain tumors, SOS1 does not appear to be mutated in human cancers, and some authors have therefore suggested that SOS1 mutations are not associated with higher risk of malignancy.34 A similar situation occurs with RAF1 mutations, which appear to be associated with cardiac abnormalities but have not been detected in somatic tumors.
LEOPARD SyndromeLEOPARD syndrome (OMIM 151100) is an autosomal dominant inherited disorder named using an acronym for the main clinical manifestations: lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retarded growth, and sensorineural deafness. This syndrome is caused by mutations in the PTP domain of PTPN11, which, unlike in Noonan syndrome, lead to loss of catalytic activity in the SHP-2 domain. It is unclear, however, how the inactivating and activating mutations in the same gene observed in LEOPARD and Noonan syndromes, respectively, can lead to similar phenotypic manifestations. In fact, patients with LEOPARD syndrome have also been described as having activating mutations in RAF1,23 making it likely that other associated factors determine the appearance of Noonan syndrome.
Affected patients have a phenotype that is very similar to that of Noonan syndrome and is accentuated with increasing age. Lentigines can appear late, and this can create difficulties for the initial differential diagnosis with Noonan syndrome. Electrocardiographic abnormalities are present in 75% of patients, and these include conduction disorders, repolarization abnormalities, and abnormal QRS complexes.35 Almost all adults have ocular hypertelorism, as well as a wide nasal bridge, palpebral ptosis, low-set ears, accentuated philtrum, and premature facial wrinkling.36 Pulmonary valve stenosis was once believed to be present in 40% of patients, but it is now thought to occur much less frequently, in around 10% to 20% of cases.35 Progressive myocardial hypertrophy is the most common heart defect and is the cause of death in many cases.36 Half of the patients have bilateral cryptorchidism and it is not uncommon to observe hypospadia, genital hypoplasia, and delayed-onset puberty.37 Around 25% of adults have lower than average height. Sensorineural deafness affects 15% to 25% of patients and, although it is usually diagnosed in infancy, can also appear late.36 If mental retardation is present, it is usually mild.
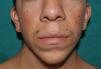
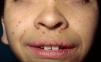
Skin ManifestationsAlthough lentigines are the most characteristic skin manifestation, they may not be present in the first few years of life,36,38 and this can cause difficulties for differential diagnosis with Noonan syndrome. These lesions are dark brown in color and affect the whole body, but they are particularly noticeable on the face and the upper body. They begin to appear at age 4 or 5 years, and their numbers increase exponentially during childhood. They are never found on the mucosas and their appearance is not related to sun exposure (Fig. 4). In around 50% of patients, café-au-lait spots are also observed and can have a typical appearance or be darker than usual (in which case they are referred to as café noir or black coffee spots), and they can appear even before lentigines.36,38 The histological appearance of the darkest spots may correspond to simple lentigines or to melanocytic nevi, and as a result some authors have proposed including melanocytic lesions within the spectrum of LEOPARD syndrome.39 Less frequently, hypopigmented lesions may occur, and cases of melanoma have also been reported.40
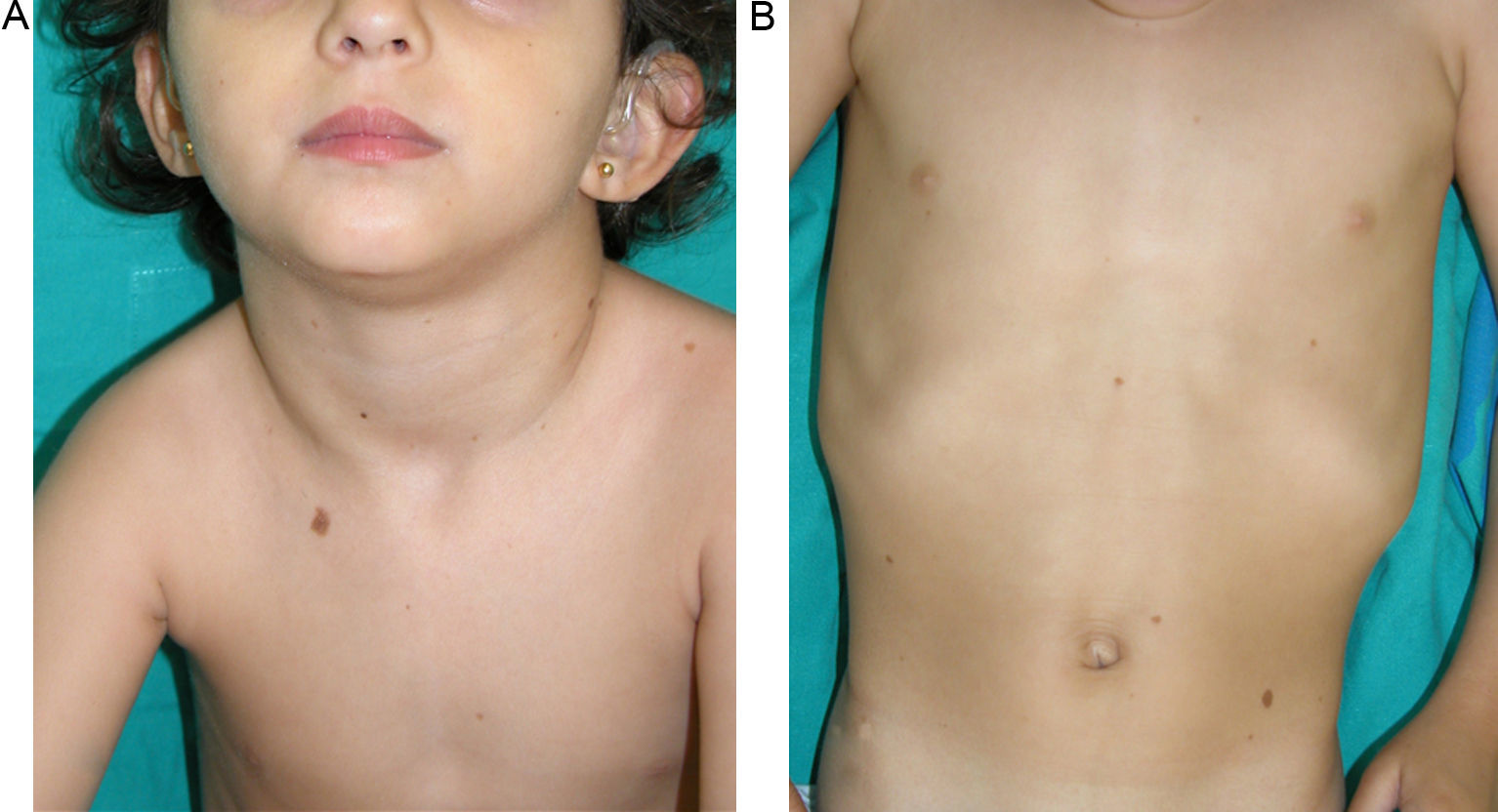
LEOPARD syndrome. A, Lentigines on the upper trunk of a girl with LEOPARD syndrome. The patient had a mutation in PTPN11 associated with deafness and ocular hypertelorism. Also note the low-set ears and wide neck, which are common phenotypic findings in Noonan syndrome. B, Lentigines on the front of the trunk (image courtesy of Dr Ana Martín Santiago).
Hematological tumors such as acute myeloid leukemia, acute lymphoblastic leukemia, and myeloproliferative disorders have been reported, along with neuroblastoma,33 bilateral choristomas, and melanoma.40
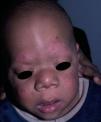
Type-1 NeurofibromatosisType-1 neurofibromatosis (OMIM 162200) is a neurocutaneous autosomal dominant inherited disorder in which 50% of cases appear spontaneously. It is caused by a mutation in NF1—the gene contains 61 exons and is located on chromosome 17q11.2, which has the highest frequency of spontaneous mutation of anywhere in the genome.41 The NF1 gene product, neurofibromin is a 327-kDa protein that functions as a negative regulator in the RAF/MAPK pathway. It contains a central domain with GAP activity that is involved in the conversion of RAS protein into the inactive GDP-bound form. When the suppressor activity of neurofibromin is reduced, the activity of the RAS/MAPK pathway increases. Most patients with neurofibromatosis have an intragenic mutation in NF1, and only 5% have a 1.4-megabase microdeletion that contains at least 11 genes.42 There is no genotype-phenotype correlation except in 2 types of genetic abnormality. First, patients with the 1.4-megabase deletion tend to be taller, have a larger number of neurofibromas, and exhibit more-pronounced learning difficulties. They have a more-pronounced facial phenotype and greater propensity to malignant tumors in the peripheral nerve bundle than those with intragenic mutations.43–45 Second, patients who have a deletion of 3 base pairs in exon 17 lack neurofibromas and have a lower frequency of serious complications.46
The expressivity of the clinical manifestations of type I neurofibromatosis is highly variable, even within the same family, and is dependent on age.47 In the 1980s, a series of diagnostic criteria were defined that included the following findings: a) 6 or more café-au-lait spots with a maximum diameter greater than 5mm in the pre-pubertal phase and greater than 15mm after puberty; b) 2 or more neurofibromas of any type or 1 plexiform neurofibroma; c) axillary or inguinal freckles; d) 2 or more Lisch nodules (hamartomas of the iris); e) optic gliomas; f) a characteristic bone lesion such as sphenoid dysplasia or thinning of the bone cortex in the long bones with or without pseudarthrosis; and g) a first-degree relative (child, sibling, or parent) who meets the above criteria. Although these criteria are fairly sensitive and specific in adults, they are less sensitive in children younger than 8 years.48 Furthermore, they do not take into account other symptoms such as learning difficulties, malignant tumors of the nerve sheath, or macrocephaly. A retrospective study published in 2000 that included 1900 cases of type-1 neurofibromatosis found that 46% of sporadic cases did not meet the diagnostic criteria in the first year of life, whereas 97% of patients met the criteria when they were 8 years old, and 100% of patients met them at age 20 years.49
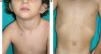
Skin ManifestationsThe most-typical dermatological findings in type-1 neurofibromatosis are included in the diagnostic criteria described above (café-au-lait spots, axillary and inguinal freckles, and neurofibromas). Café-au-lait spots appear during the first year of life in 99% of patients and usually increase in number in the course of childhood. Although these spots can occur in healthy patients without type-1 neurofibromatosis, it is calculated that less than 1% of healthy children younger than 5 years have more than 2 café-au-lait spots.50 The café-au-lait spots are not found on the palms of hands, soles of the feet, or scalp, and vary in size even within the same patient.51 They generally clear over time and do not create cosmetic problems. Axillary and inguinal freckles (traditionally known as the Crowe sign) appear between age 3 and 5 and are considered to be the most-specific skin finding; some authors consider them to be almost pathognomonic.48 Almost 90% of affected adults have freckles, and these are often not restricted to the skin folds but rather extend throughout the trunk, neck, and even around the lips52 (Fig. 5). Neurofibromas can appear on any part of the body but do so at a later stage, generally after puberty. Their size is variable and they are often the main complaint due to their volume or visibility. Superficial plexiform neurofibromas are a particular subtype of neurofibromas that tend to be congenital; they are hyperpigmented and exhibit hypertrichosis and are therefore often confused with congenital melanocytic nevi (Fig. 6). Rarer variants of type-1 neurofibromatosis include reddish-blue and pseudoatrophic macules (Fig. 7), which are lesions with a peculiar appearance related to the presence of neurofibromatous tissue infiltrating the dermal vessels or surrounding them, respectively.53 Less-characteristic cutaneous findings that are nevertheless relatively frequent in type-1 neurofibromatosis include juvenile xanthogranulomas, glomus tumors, melanomas, pale nevi, generalized hyperpigmentation, and itching.48
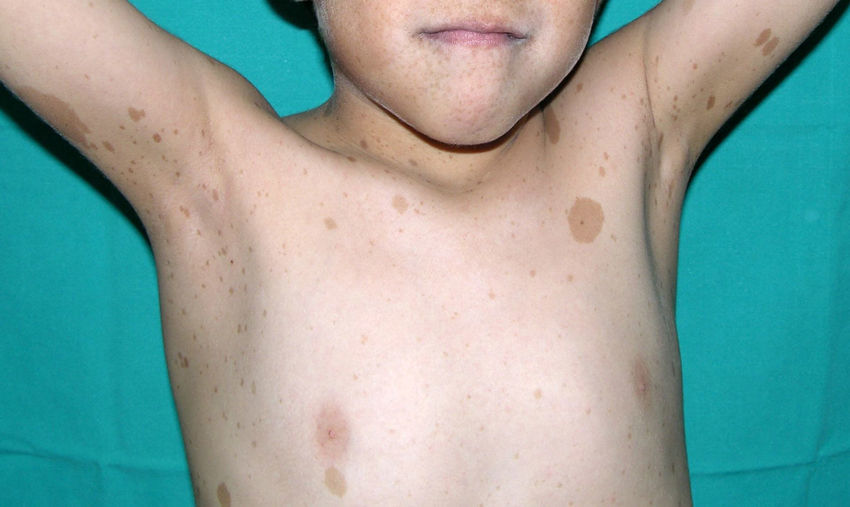
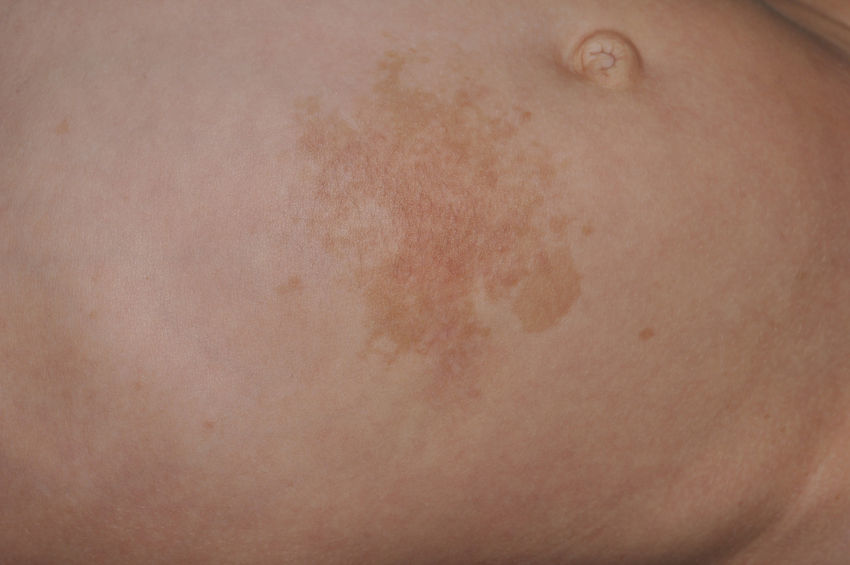
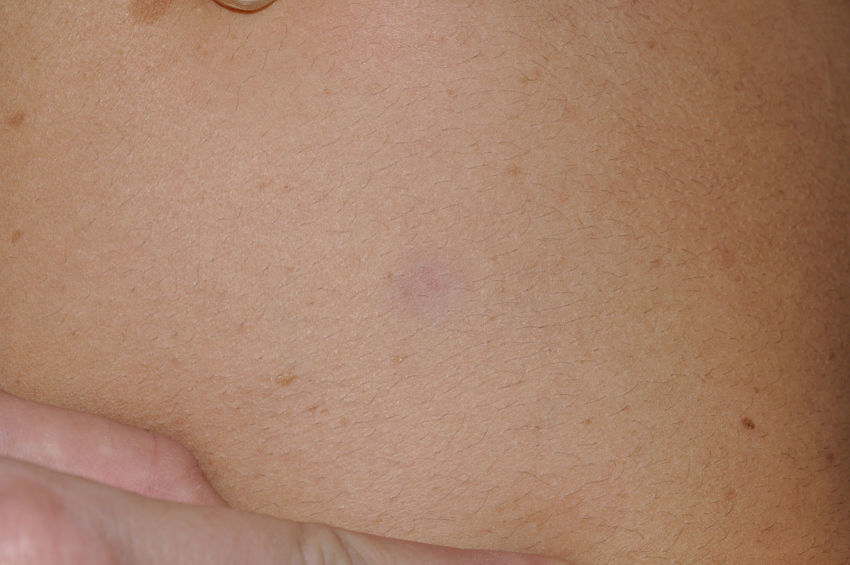
There is a mosaic form of type-1 neurofibromatosis known as segmental neurofibromatosis, which is characterized by involvement of a segment of the body and the absence of a family history of the disease. Isolated café-au-lait spots, isolated neurofibromas, simultaneous occurrence of café-au-lait spots and neurofibromas, and isolated plexiform neurofibromas may all be observed.54 The risk of passing on the disease is unknown, but transmission does occur if the condition coexists with gonadal mosaicism.55 The risk does not depend on the affected site, and therefore the possibility of transmission exists even when the segmental form does not involve the genital region.54
Type-1 Neurofibromatosis and Risk of CancerAll patients with type-1 neurofibromatosis have a greater tendency to develop either malignant or benign tumors (such as neurofibromas or glomus tumors). Between 8% and 12% of patients with type-1 neurofibromatosis develop malignant tumors of the peripheral nerve bundle, generally in the region of a plexiform neurofibroma.56 Persistent pain and rapid growth should be considered warning signs, and local itching could indicate the presence of a tumor in the central nervous system.57 In these patients there are also higher rates of sarcoma,58 rhabdomyosarcoma, neuroblastoma, gastrointestinal stromal tumors, pheochromocytoma, and breast cancer.59 Finally, children with type-1 neurofibromatosis have a risk of juvenile myelomonocytic leukemia that is 200 to 500 times higher than in the healthy population, independently of the presence or absence of juvenile xanthogranulomas60; Nevertheless, screening for juvenile myelomonocytic leukemia is not advised except in the presence of suggestive additional findings such as hepatosplenomegaly, enlarged lymph nodes, pallor, or petechiae.
Legius SyndromeLegius syndrome (OMIM 611431), or type-1 neurofibromatosis-like syndrome is a syndrome similar to type-1 neurofibromatosis. Described in 2007,61 this autosomal dominant inherited disease is caused by a mutation in SPRED1 (sprouty related EVH1 domain-containing protein 1), a gene containing 7 exons located on chromosome 15q13.2 that, when mutated, results in a loss of repressor function on RAF.62 Consequently, there is an increase in the level of RAF1, MEK, and ERK proteins along with an increase in the transcription factor ELK1. Mutations have been detected in all of the exons of the gene but, while some of them are recurrent, there are no recognized mutation hotspots.63 Legius syndrome is characterized by the presence of café-au-lait spots or lentigines, occasionally associated with macrocephaly, a Noonan-type phenotype, and/or learning difficulties. These patients often meet the criteria for type-1 neurofibromatosis, but in Legius syndrome the presence of Lisch nodules, neurofibromas, tumors of the central nervous system, and mutations in NF1 are specifically excluded.61
Skin ManifestationsThe typical cutaneous findings observed in Legius syndrome include café-au-lait spots and lentigines. Some adults also have lipomas.62,63 Published series suggest familial cases with café-au-lait spots with or without lentigines are the most likely to have Legius syndrome.63
Legius Syndrome and Risk of CancerIt is not clear whether Legius syndrome is associated with a predisposition towards cancer. Patients have been described with acute monoblastic leukemia,64 lung cancer, Wilms tumors, adenocarcinoma of the colon,61 breast cancer, ovarian dermoid tumor,65 and vestibular Schwannoma.63 The possibility of increased risk of tumors must therefore be taken into account.
Costello SyndromeCostello syndrome (OMIM 218040) is a sporadic condition that appears to display autosomal dominant inheritance. The causative gene was identified in 2005.66 This genetic condition was the first to be identified as a consequence of a germline mutation in the RAS pathway. Almost all patients with Costello syndrome carry mutations in the gene encoding HRAS.67 The gene is located on chromosome 11p15.5, contains 6 exons, and is 6.5 kilobases long4 and encodes 2 different proteins, p21-HRAS and p19-HRAS; p21-HRAS is much more abundant than 19p-HRAS, which appears to exert a negative regulatory effect on p21-HRAS.68,69 The mutation in HRAS leads to an abnormal activation of the RAS pathway. Almost all patients have a heterozygous mutation on the paternal chromosome, but isolated cases exist in which the abnormal chromosome is inherited from the mother,70,71 and some exceptional cases of mosaicism have been reported.72,73 In a series of 139 cases, the pG12S mutation was present in up to 80% of cases and the pG12A mutation in another 7%.74 Consequently, genetic analyses should start with exon 2, where the mutation is located in the majority of cases. In the remaining patients, other mutations were found, with the same mutation never identified in more than 4 patients. The most common mutations give rise to a typical phenotype, whereas other mutations such as pT85I or pG12V are associated with a milder75 or stronger66,76 phenotype, respectively. The fact that all patients carry heterozygous mutations indicates an autosomal dominant pattern of inheritance; all of the cases reported to date occurred de novo in the parents, with a mean age of between 38 and 43 years.77,78 A phenotype corresponding to Costello syndrome has also been reported in patients with mutations in KRAS79,80 and in a patient with a mutation in BRAF.81 These reports reveal the wide phenotypic spectrum of patients with mutations in genes for the RAS pathway.
The gestation of these patients is associated with polyhydramnios (90%), older parental age (62%), macrosomia (50%), premature birth (50%), and, less frequently, fetal tachyarrhythmia.74 Notable feeding difficulties, usually requiring a nasogastric tube, are observed during the postnatal period. Over time, weight gain continues to be delayed and, although the cephalic diameter is within normal limits, patients have a macrocephalic appearance. General developmental delay and hypotonia are apparent in the first few months of life and lead to a moderate cognitive deficit (IQ, 56-59) when the child is older and to a particularly clear deficit in facial expressiveness. The sociability of the patients is very noticeable, and they often interact with the physician by asking questions and making comments. The dysmorphic facial features are accentuated over time and include a large mouth with thick lips, chubby cheeks, a prominent forehead, low hairline, epicanthus, short nose with a depressed nasal bridge, and low-set, posteriorly rotated ears with a thickened helix and lobes (Fig. 8).82 Cubital deviation of the wrist and thumb are highly characteristic, as are retraction of the heel (almost invariant), kyphoscoliosis, and osteopenia. Growth hormone deficiencies are common, as are dysregulation of pubertal development. At least two thirds of patients have some type of heart defect, including cardiac hypertrophy (41%), congenital heart defects (21%), and supraventricular tachycardia (33%) among the most common findings.83
Skin Manifestations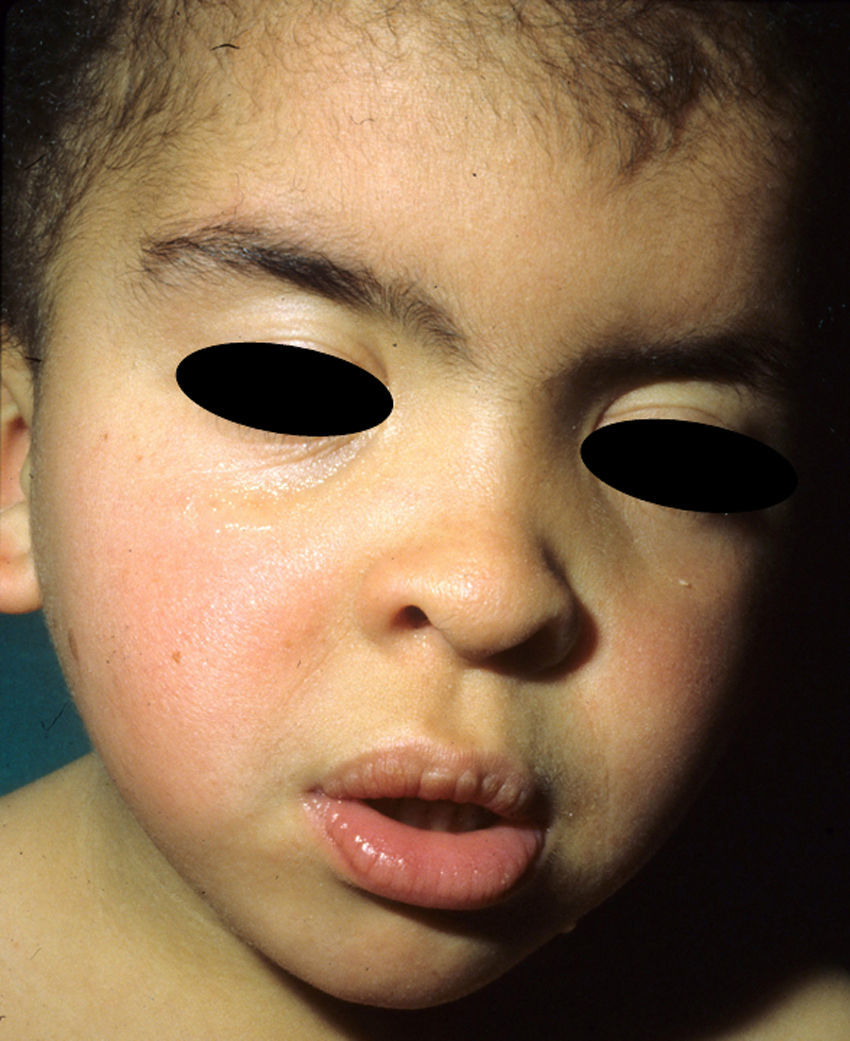
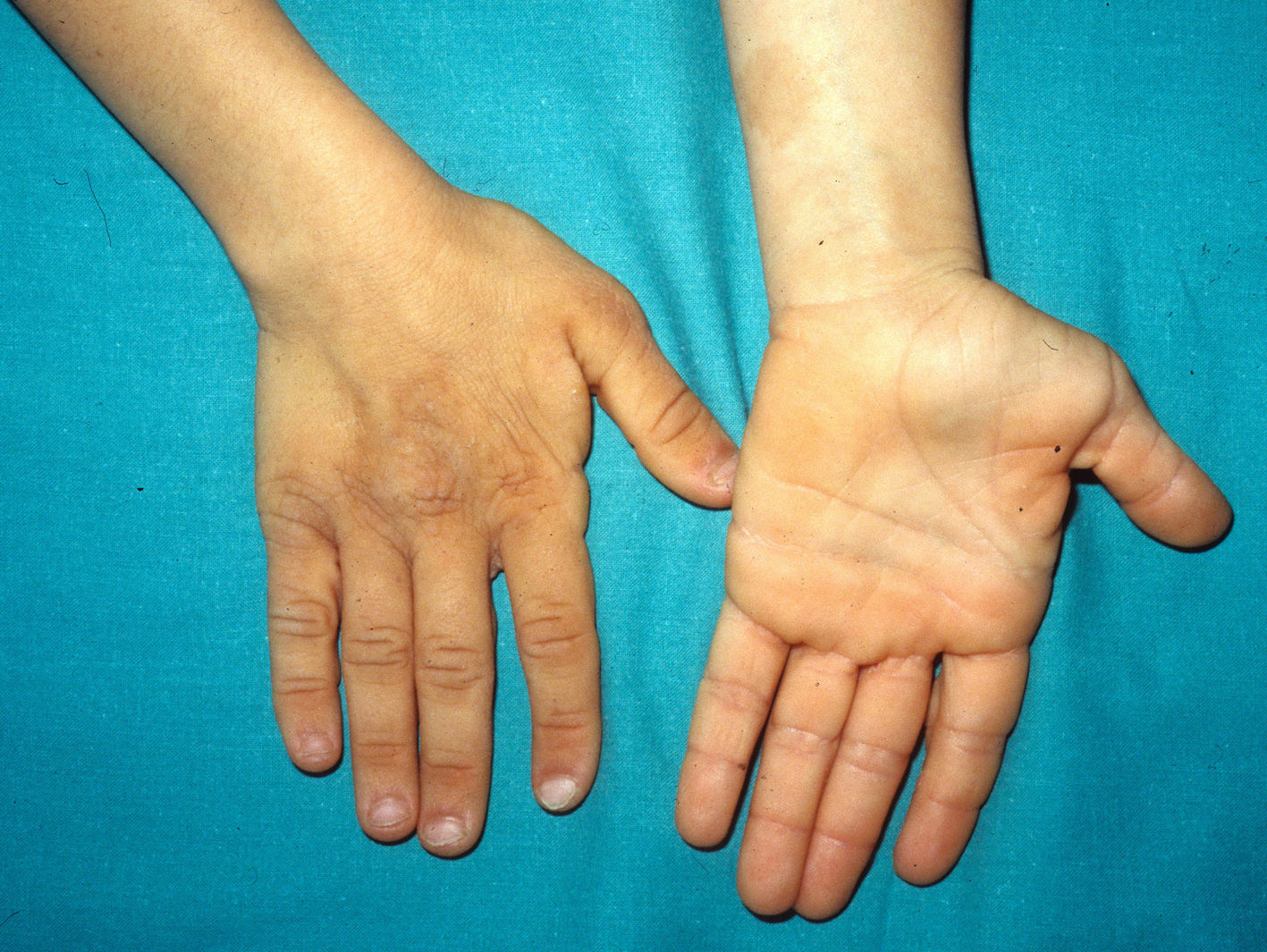
The most significant, although not pathognomonic, cutaneous findings are short, thin, curly hair, papillomatous lesions around the mouth and nostrils (Fig. 9), hyperpigmentation of skin folds, slack skin, redundant skin on the dorsum of the hands and feet, and marked accentuation of lines on the palms of the hands and soles of the feet (Fig. 10). Papillomatous lesions can appear around the orifices during the first year of life or later during adolescence.82 In these patients, histology reveals more fragmentation and anastomosis of elastic fibres than is usual in normal healthy skin.84 The fibroblasts in these patients appear to have a functional deficiency of the elastin-binding protein that leads to abnormalities in the elastic fibers and abnormal deposition of the fibers in the skin, tongue, pharynx, larynx, alveoli, and aorta.85,86
Costello Syndrome and Risk of Cancer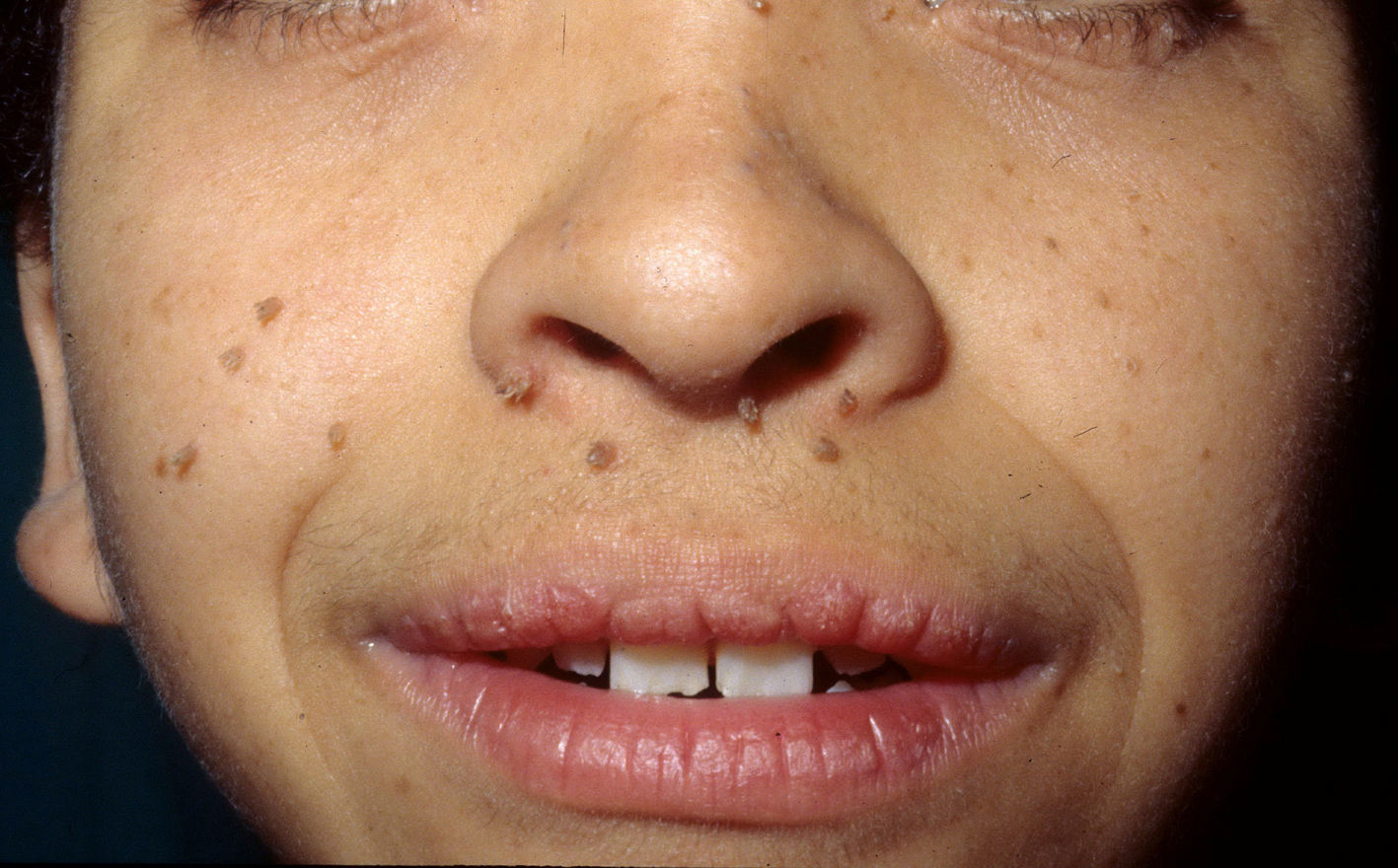
Patients with Costello syndrome have an increased tendency to develop both benign and malignant tumors. Between 13% and 15% of patients will develop cancer during their lifetime.20,74 Embryonic rhabdomyosarcoma is the most common tumor, followed by neuroblastoma, transitional cell carcinoma of the bladder, and other less frequent cancers, such as adenocarcinoma of the bladder or acoustic neuroma.87 Rhabdomyosarcoma and neuroblastoma appear during childhood, generally before the age of 4 years, whereas bladder cancer is more common in adults, although it can appear at any time after 10 years of age.87,88 Interestingly, leukemia has not been reported in any of these patients.33 Given the evidence of increased risk of malignancy, follow-up is advisable.88 The likelihood of cancer appears to be greater in patients with the G12A mutation than in those with the G12S mutation.20
Cardiofaciocutaneous SyndromeThe cardiofaciocutaneous syndrome (OMIM 115150) is a rare sporadic genetic disorder that displays autosomal dominant inheritance. To date, it has been linked to mutations in BRAF, MAP1K1, MAP2K2, and KRAS. BRAF is located on chromosome 7q34, contains 18 exons, and is 190 megabases long. Between 40% and 78% of patients with cardiofaciocutaneous syndrome carry germline mutations in BRAF,89,90 which is almost never mutated in other RASopathies. The most common mutation is Q257R, which is located on exon 6, followed by other mutations in exons 11, 12, 14, and 15. Approximately 25% of patients have a mutation in MAP2K1 or MAP2K2,19 which are located on chromosomes 15q22 and 19p13.3, respectively. Both contain 11 exons and encoded the proteins MEK1 and MEK2, respectively. These proteins are activated by RAF and can phosphorylate ERK1 and ERK2. KRAS is located on chromosome 12p12.1, contains 6 exons, and produces 2 mRNA isoforms, one of which (KRASA4a) is expressed ubiquitously and the other (KRASA4b) is not expressed in the heart.91 Less than 1% of patients carry mutations in this gene, the most frequent being D153V; this mutation affects the generation of the KRASA4b isoform, suggesting that this isoform participates in human development.20 Some authors have suggested that there is no clear genotype-phenotype relationship, 92 whereas others have suggested that such a relationship does indeed exist. For instance, KRAS mutations are associated with a lower frequency of skin manifestations,90 whereas patients with mutations in MEK display normal cognitive development.11 Germline mutations in KRAS have been described in patients with cardiofaciocutaneous syndrome, Noonan syndrome, and Costello syndrome, and in some patients with combined Noonan syndrome and cardiofaciocutaneous syndrome phenotype or a combined Costello syndrome and Noonan syndrome phenotype, revealing once again the degree of clinical overlap among these patients.93
The phenotypic findings often overlap with those of other RASopathies, particularly Noonan syndrome. The clinical features are so similar in some patients that for some time the cardiofaciocutaneous syndrome was considered to be a severe form of Noonan syndrome; nevertheless, the substantial mental retardation and ectodermal cutaneous abnormalities are clinically suggestive of cardiofaciocutaneous syndrome. Additionally, genetic studies facilitate a definitive diagnosis, since cardiofaciocutaneous syndrome is never associated with mutations in PTPN11. Polyhydramnios usually occurs during the gestation of these patients and there is developmental delay in the postnatal period caused by feeding difficulties. Dysmorphic facial features include a wide forehead with bilateral temporal constriction, downward curving splits in the eyelids, depressed nasal bridge, and posteriorly rotated ears with a prominent helix. Psychomotor retardation is moderate or severe, and 75% of patients usually display one or more cardiac defects, usually pulmonary stenosis (45%), myocardial hypertrophy (40%), and atrial septal defects (22%).93
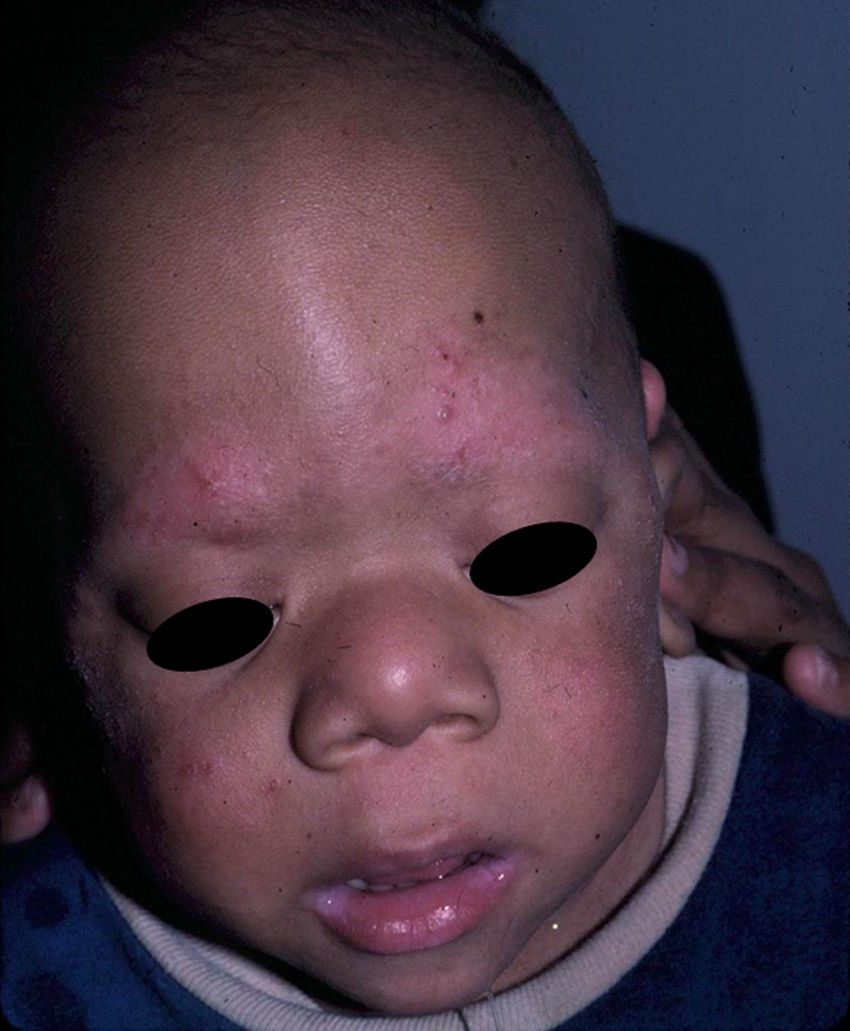
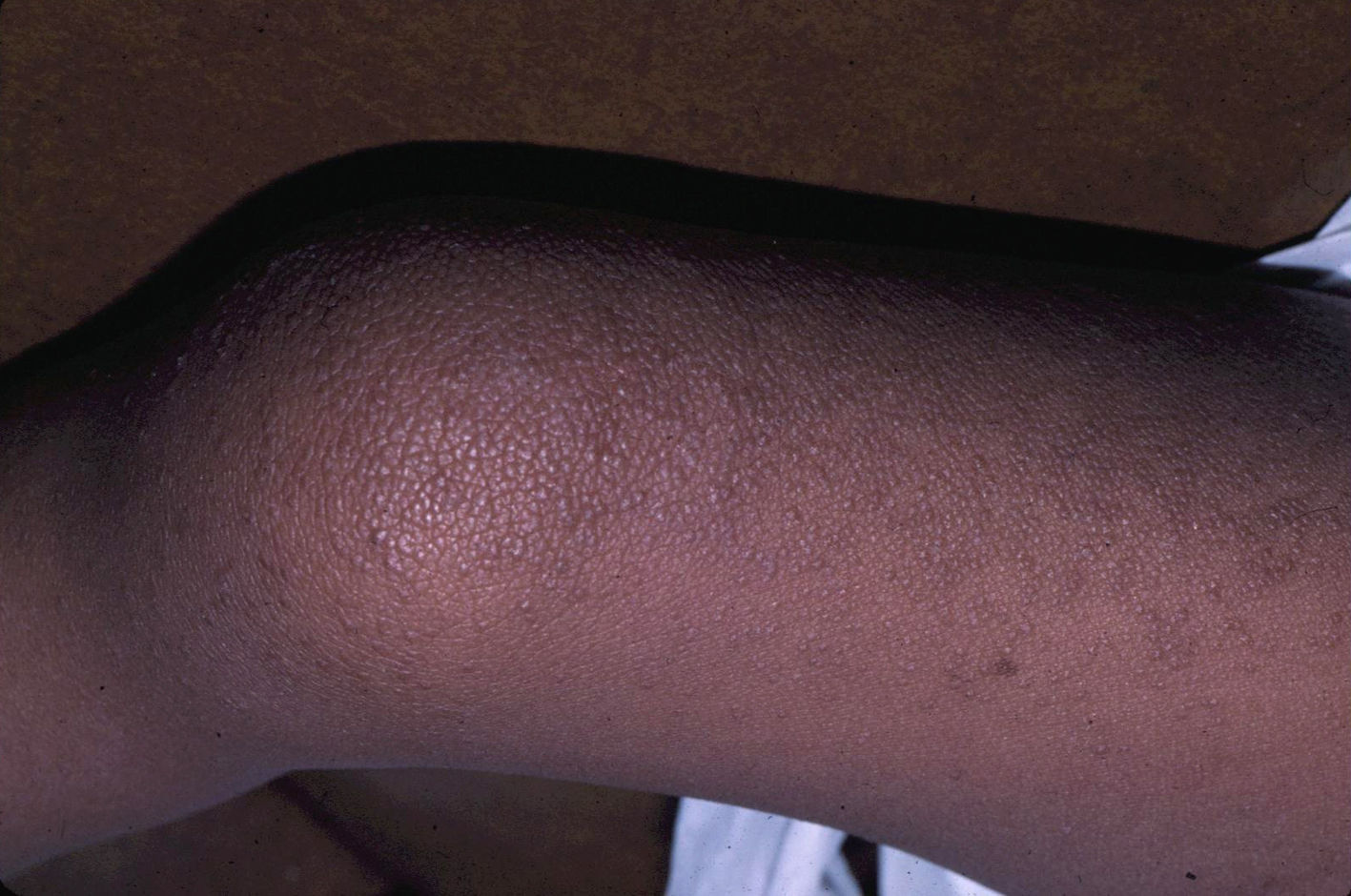
Skin ManifestationsSubstantial ectodermal abnormalities are observed in cardiofaciocutaneous syndrome and are often helpful in the differential diagnosis. The patients have short, thin, curly hair, ichthyosiform scaling, and generalized follicular hyperkeratosis (Fig. 11). A distinctive finding is ulerythema ophryogenes with follicular hyperkeratosis, erythema, and scarring alopecia of the eyebrows (Fig. 12). When affected individuals reach adulthood, palmoplantar hyperkeratosis and lymphedema may appear. In a series of 61 patients published in 2010, hair abnormalities were observed in 93% of patients, ulerythema ophryogenes in 90%, keratosis pilaris in 80%, palmoplantar hyperkeratosis in 36%, more than 50 acquired melanocytic nevi in 23%, and childhood hemangiomas in 26%.94 Other cutaneous findings (such as rapid growth of the nails, scarcity of body hair on the limbs, odor from the armpits prior to puberty, scarce hair growth, folding of the earlobes, acanthosis nigricans, and hyperplastic nipples) were also reported. Although in that series, no patients had café-au-lait spots or papillomas around the orifices, findings which are common to other RASopathies, other authors have reported patients with café-au-lait spots and lentigines.17,95 Finally, some authors have highlighted the possibility of affected sweat ducts and hair follicles, since histology in 1 patient revealed squamous eccrine metaplasia and periadnexal granulomas.96
Unlike other syndromes involving the RAS/MAPK pathway, cardiofaciocutaneous syndrome appears to confer no increased risk of malignancy. However, 2 patients have been described with germline mutations in BRAF who developed acute lymphoblastic leukemia,90,97,98 another with metastatic hepatoblastoma,99 and another with neuroblastoma.90 The potential risk should therefore not be ignored.
Autoimmune Lymphoproliferative SyndromeAutoimmune lymphoproliferative syndrome (OMIM 164790) is characterized by a defect in lymphocyte apoptosis that leads to the accumulation of normal lymphocytes in the organism and an increased risk of hematological tumors.100 Most cases are due to a defect in apoptosis mediated by the cell-surface Fas receptor caused by mutations in the CD95 pathway,101 but recently germline mutations in NRAS have been identified that lead to the same disease via an independent mechanism.12 The mutation in NRAS leads to stabilization of the active GTP-bound form, which results in activation of the entire RAS pathway. Thus, phosphorylation of ERK is increased, leading to inhibition of lymphocyte expression of BIN and, consequently, intrinsic mitochondrial apoptosis.
Capillary Malformation–Arteriovenous Malformation SyndromeCapillary malformation-arteriovenous malformation syndrome (OMIM 608354) is an autosomal dominant condition in which multifocal capillary malformations and arteriovenous fistulas form in the skin, muscles, bones, and internal organs such as the brain and heart.13,102 It is caused by inactivating mutations in RASA1, which like NFI encodes a RAS-GAP protein; insufficiency of this protein leads to a reduction in the hydrolysis of RAS-GTP and consequent overstimulation of the RAS/MAPK pathway.102 Whether these patients have an increased risk of developing tumors is not known.
Hereditary Gingival FibromatosisHereditary gingival fibromatosis (OMIM 135300) is characterized by slow, progressive, benign fibrous growth of the gums. It is a genetically heterogeneous condition that can display both autosomal dominant and recessive inheritance. Type-1 hereditary gingival fibromatosis is a rare dominant form that is caused by a mutation in SOS1 that activates the RAS/MAPK pathway.14 It is not clear why there are no other associated developmental defects, as occurs with other mutations in SOS1 that are linked to Noonan syndrome.
RAS and CancerAround 30% of human tumors carry activating mutations in the RAS/MAPK pathway.6 The prevalence varies according to the tumor: 90% in pancreatic adenocarcinoma, 50% in cancer of the colon and thyroid gland, 30% in lung cancer, and 25% in melanoma. Mutations are detected most frequently in KRAS (around 85% of all cases) and NRAS (around 15%), and much less frequently in HRAS (less than 1% of the total). These 3 members of the RAS family share 85% amino acid sequence homology and are expressed in many tissues in the organism, especially in the case of KRAS, which is detectable in almost all cell types.103 Mutation hotspots in the RAS proteins lead to changes in amino acids G12, G13, and Q61, which block both intrinsic hydrolysis of the active form of RAS bound to GTP, and the response to GAPs. This leads to an unlimited intrinsic activation of the MAPK pathway, of which the RAF protein is an effector and a known proto-oncogene. There are 3 isoforms of RAF, of which BRAF, the most effective of those within the MPAK pathway, is involved in 7% of all human cancers and is particularly common in melanoma, where it is found in 70% of cases.104 Most of the mutations occur within the kinase domain, and the most frequent one leads to a V600E substitution that activates the RAS/MAPK pathway.105 Finally, somatic mutations have been observed in PTPN11 in patients with juvenile myelomonocytic leukemia, acute lymphoblastic leukemia, and acute myeloid leukemia; PTPN11 is therefore considered an oncogene.106
Future Perspectives in DermatologyThe RAS/MAPK pathway is well defined, and abnormalities in the pathway affect the development of cancer and the appearance of complex genetic syndromes, as discussed in this review. Since the discovery of the role of this pathway in tumor biology 30 years ago, research has sought antitumor drugs to block it. Unfortunately, these efforts have been largely unsuccessful so far. In dermatology, research is mainly focused on melanoma, the tumor in which mutations in BRAF and NRAS are found in 50% to 70% and 15% to 30% of cases, respectively,107,108 and for which few therapeutic options are available. BRAF and MEK proteins would appear to be the ideal targets within this pathway, but it is also necessary to inhibit other metabolic pathways—such as the AKT3 and PI3K pathways—that are involved in development, growth, and spread of melanoma.109 Sorafenib, a RAF inhibitor approved for the treatment of advanced hepatocarcinoma, has not been found to be effective in the treatment of melanomas with BRAF V600E mutations,110 probably due to feedback loops that maintain hyperactivation of the pathway. Drugs that inhibit MEK and ERK appear to be more promising, but as yet they do not have bioavailability and toxicity profiles that would allow their use.109
Clinical trials are underway to attempt to delay the development of tumors and improve cognitive development in patients with type-1 neurofibromatosis. Studies have been initiated with sirolimus (an inhibitor of the mTOR pathway that is dysregulated in types 1 and 2 neurofibromatosis and tuberous sclerosis), imatinin, cediranib (an inhibitor of VEGFR, c-Kit, and other serine proteases), sorafenib, bevacizumab combined with RAD001, and lovastatin.111 It remains unclear whether these drugs will be able to compensate for the feedback mechanisms of the inhibited proteins; nor do we yet know whether the long-term side effects will be balanced by the hypothetical benefits of their use. Undoubtedly, the low incidence of RASopathies reduces the opportunities available to include patients in appropriate trials and diminishes the financial incentive for research into their treatment. Consequently, patients may have to wait some time before they can benefit from pharmacological therapy.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Hernández-Martín A, Torrelo A. Rasopatías: trastornos del desarrollo con predisposición al cáncer y manifestaciones cutáneas. Actas Dermosifiliograf. 2011;102:402-416.


