





Lymphomatoid papulosis (LyP) is a CD30+ lymphoproliferative skin disease that has been described in association with Hodgkin lymphoma. It has also been reported to progress to mycosis fungoides or cutaneous anaplastic large-cell lymphoma.
ObjectiveTo study the clinical and histologic features of LyP and response to treatment in a patient series.
Materials and methodsFor this retrospective, descriptive, observational study of patients with histologically confirmed LyP and sufficient follow-up data on record, we extracted histologic findings on skin biopsy, clinical presentation, clinical course, and response to treatments.
ResultsEighteen patients (10 male, 8 female) were identified. Most biopsies (14/18, 78%) showed a wedge-shaped lymphocytic infiltrate with CD30+, CD3+, and CD56− cells. A type A histologic pattern was present in the biopsies of 83% of the patients. The most common presentation (83%) consisted of papules on the trunk; for 62% LyP resolved after a single episode. Twelve percent of the patients developed mycosis fungoides (mean follow-up, 7 years); no other associations were noted.
DiscussionAlthough few series of patients with LyP have been published in recent years, the findings reported generally coincide with our observations.
ConclusionLyP is typically a CD30+ lymphoproliferative disorder that usually runs a benign course and responds well to treatment.
La papulosis linfomatoide (PL) es una dermatosis que se engloba dentro de los procesos linfoproliferativos CD30 positivos de la piel. Se ha descrito su asociación a linfoma de Hogking (LH), así como su progresión a micosis fungoide (MF) y linfoma cutáneo anaplásico de célula grande (LCACG).
ObjetivosInvestigar los hallazgos clínicos, histológicos y la respuesta al tratamiento en un grupo de pacientes con PL.
Material y métodosSe llevó a cabo un estudio retrospectivo, descriptivo y observacional. Se seleccionaron 18 pacientes con diagnóstico histológico confirmado de PL y con un adecuado seguimiento clínico. Se recopilaron los hallazgos histológicos de las biopsias de piel, la forma de presentación, la evolución y la respuesta a los tratamientos utilizados.
ResultadosSe reclutaron un total de 18 pacientes, 10 varones y 8 mujeres. La mayoría de las biopsias, 14 de 18 (78%) mostraban un infiltrado linfocitario en cuña, CD30 positivo, CD3 positivo y CD56 negativo. El tipo histológico más frecuente fue el tipo A, presente en un 83% de las biopsias de los pacientes. La forma clínica de presentación más frecuente fue en forma de pápulas en el tronco (83%). Un 62% de los pacientes sufrió un único brote autorresolutivo. La media de seguimiento fue de 7 años, durante los cuales un 12% de los pacientes desarrolló una micosis fungoide, sin encontrarse otras asociaciones.
DiscusiónExisten pocas series de pacientes con PL publicadas en los últimos años; sin embargo, globalmente los hallazgos descritos en ellas coinciden con las de nuestro grupo de pacientes.
ConclusionesLa PL es un cuadro linfoproliferativo típicamente CD30 positivo que habitualmente tiene un curso benigno con buena respuesta a los tratamientos utilizados.
Lymphomatoid papulosis (LyP) is a lymphoproliferative disorder that affects middle-aged patients in the form of recurrent outbreaks of papules or papulonecrotic lesions. It runs a benign course and usually resolves spontaneously in 4 to 6 weeks. However, as it can progress to other types of cutaneous T-cell lymphoma and is associated with Hodgkin lymphoma, patients should be monitored. Because the course of LyP is self-limiting and benign, treatment is often not prescribed or is restricted to topical corticosteroids until the lesions have resolved. When the lesions are more protracted or more extensive, they can be treated using psoralen–UV-A, low-dose methotrexate, or interferon alfa.1–5
LyP has traditionally been considered a primary cutaneous CD30+ lymphoproliferative disorder and is classified into 3 histologic patterns: type A (histiocytoid), which is characterized by the presence of large atypical lymphocytes accompanied by small lymphocytes, neutrophils, histiocytes, and eosinophils; type B (mycosis fungoides–like), which comprises a monomorphic infiltrate of small to medium-sized lymphocytes with cerebriform nuclei; and type C, which is formed by an infiltrate of large lymphocytes similar to that of type A, but in which the lymphocytes account for more than 50% of the infiltrate, thus mimicking cutaneous anaplastic large-cell lymphoma. Given the histologic overlap between LyP and other cutaneous lymphoproliferative disorders and the potential of this condition to evolve to or be associated with malignancy, the debate over whether it is benign, premalignant, or malignant remains unresolved. However, LyP is now widely accepted as a primary cutaneous lymphoma with a favorable prognosis. Given the absence of molecular or immunohistochemistry prognostic criteria, the correlation between clinical and histologic features and patient follow-up is essential for predicting disease course.1,6
Clonal rearrangement of T-cell receptors is observed in up to 60% of LyP lesions, with the same clone being found in separate lesions7,8; nevertheless, some authors have observed this clonality in small lymphocytes, thus preventing differentiation from reactive conditions.9 The multiple myeloma oncogene 1 marker was recently suggested to be a differentiating feature between LyP and cutaneous anaplastic large-cell lymphoma10; however, this role was rejected in later studies.11
The literature contains few LyP series and even fewer studies that contrast clinical data with histology and immunohistochemistry findings.
ObjectivesOur objectives were to investigate histology and immunohistochemistry findings in skin biopsies from a group of patients with LyP and to describe the clinical presentation, course of the disease, association with other conditions, response to treatment, and potential correlations between these variables and histology findings.
Material and MethodsWe performed a retrospective, descriptive, observational study of patients with a clinically and histologically confirmed diagnosis of LyP and with sufficient follow-up data on record. We selected patients using lymphomatoid papulosis as a search term in the histopathology database, which covers the period January 2000 to May 2010, and in the dermatology database, which covers the period January 1995 to May 2010. The databases yielded 44 and 24 patients, respectively. After ruling out patients who did not have an accurate diagnosis, a clinical history, or histology samples and those who were lost to follow-up, we identified 18 patients and 26 biopsy specimens.
We evaluated the following clinical variables: age at onset, sex, clinical presentation (type of lesions and site), laboratory results at diagnosis, the course of the lesions, treatments received, and the response to treatment. We also described associations with other lymphomas (both cutaneous and noncutaneous) and the latency period until these associations developed. As for histology findings, we studied type of LyP (A, B, C, or mixed), type of infiltrate (wedge-shaped or band-like), presence of epidermotropism, and positivity for CD30 (activated T-cell and B-cell marker and Hodgkin lymphoma cell marker), CD3 (T-cell marker), and CD56 (natural killer cell marker), as well as the intensity of these markers. We also determined the presence of clonal T-cell receptor rearrangement in patients who underwent testing. Immunohistochemistry markers were classified into 5 degrees of intensity (+++, ++, +, +–, and –) based on the consensus reached by 2 observers. These markers were tested using the following reagents:
- -
CD3: rabbit polyclonal antibody (Dako) IR503, Flex
- -
CD30: mouse polyclonal antibody (Dako) IR602, Flex
- -
CD56: synthetic monoclonal antibody diluted 1/25 (Master Diagnostics) Clone IB6
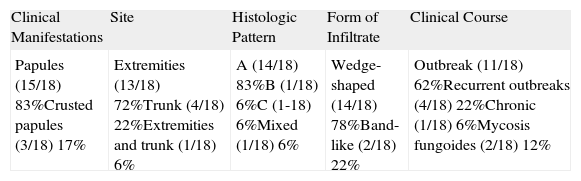
The results are summarized in Tables 1 and 2. The study sample comprised 18 patients (10 men and 8 women) with a mean age of 42.7 years (range, 7-70 years). Onset was mainly in the form of papules (15 patients [83%]) and crusted papules (3 patients [17%]) (Fig. 1). The most frequent locations were the extremities (13 patients [72%]), trunk (4 patients [22%]), and both (1 patient [6%]).
Summary of Clinical and Histologic Findings.
| Clinical Manifestations | Site | Histologic Pattern | Form of Infiltrate | Clinical Course |
| Papules (15/18) 83%Crusted papules (3/18) 17% | Extremities (13/18) 72%Trunk (4/18) 22%Extremities and trunk (1/18) 6% | A (14/18) 83%B (1/18) 6%C (1-18) 6%Mixed (1/18) 6% | Wedge-shaped (14/18) 78%Band-like (2/18) 22% | Outbreak (11/18) 62%Recurrent outbreaks (4/18) 22%Chronic (1/18) 6%Mycosis fungoides (2/18) 12% |

The results of a complete blood count and biochemistry (including lactate dehydrogenase) at diagnosis were normal for all patients, as were the results for β2-microglobulin in the 5 patients who were tested for it. Mean follow-up after diagnosis was 7 years (range, 2-22 years). The majority of patients (11 patients, 62%) had a single outbreak that resolved without treatment or with topical treatment, 4 patients (22%) experienced recurrent outbreaks, 2 patients (12%) developed associated mycosis fungoides or had both types of cutaneous T-cell lymphoma as their presenting complaint, and 1 patient continued to be affected by the chronic form, with no response to treatment. Patients who experienced recurrent outbreaks over the years responded well to psoralen–UV-A during each outbreak. In the patient with chronic disease and the patient with mycosis fungoides presenting simultaneously with LyP, psoralen–UV-A combined with methotrexate partially controlled the lesions.
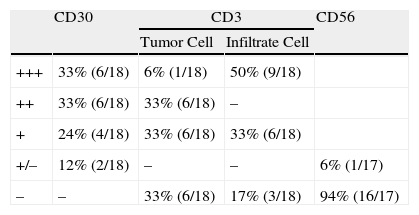
As biopsy had been repeated in some cases during follow-up, 26 specimens were available for the 18 patients. Wedge-shaped infiltrate was more common than band-like infiltrate (78% vs 22%). The most common histologic pattern was type A (15 patients [83%]) (Fig. 2); types B, C, and mixed AB were found in 1 patient each. All the samples were positive for CD30; in most cases this positivity was moderate or intense (Fig. 3). Positivity for CD3 was more intense in the cells of the infiltrate than in the tumor cells (Fig. 4), which were also positive for CD3, although less intensely. Interestingly, among the cells accompanying the tumor cells, one group of cells was positive for CD3 while the other was not, as if the cell populations were different (even though both had similar morphology). None of the cells were positive for CD56, although in 1 case this finding was doubtful (Fig. 5). Epidermotropism was only found in type B and mixed AB. T-cell receptor rearrangement was not detected in the 9 patients who underwent testing. Curiously, findings in repeat biopsy specimens did not vary (3 patients and 7 specimens).
.")
.")
.")
.")
Table 1 summarizes the clinical characteristics of the patients and histologic patterns of LyP; no apparent correlation was found between the clinical variables studied and the histologic type. The small sample size prevented us from applying statistical tests with sufficient power to be able to analyze the variables described.
DiscussionThe epidemiologic and clinical findings for our group of patients are consistent with those published in the literature,2,3 that is, LyP is a lymphoproliferative disease that is slightly more common in men and has a mean age at onset of 40 to 45 years. In most cases, onset takes the form of papules or crusted papules on the limbs; however, several forms have been reported (eg, vesicular, agminated, and ulcerative-necrotic).1
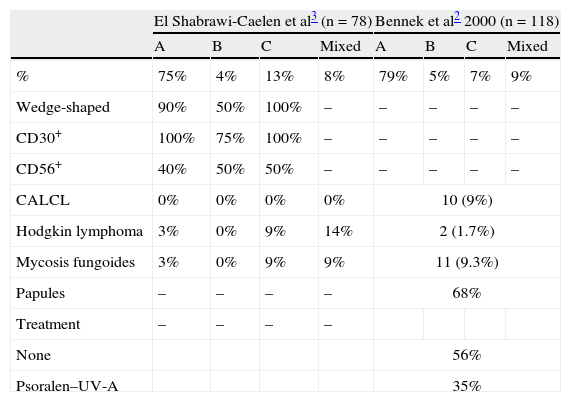
A review of the literature from the last 10 years revealed only 2 series that analyze clinical and histologic findings and their association with other diseases in patients with LyP (Table 3)2,3. In addition, the Spanish medical literature includes a recently published series comprising 9 pediatric patients with LyP.4 Data on the histology of the lesions reported by El Shabrawi-Caelen et al3 in 2004 are consistent with our results, namely, a clear predominance of type A (75% of patients). Types B and C were less common, with a frequency of 4% and 13%, respectively. Again, these findings were consistent with those of our series, in which each type affected 6% of patients. Most infiltrates were wedge-shaped, except for those of patients with type B disease, which, given their resemblance to the infiltrates occurring in mycosis fungoides, tended to be band-like. Similarly, in 2000, Bennek et al2 reported on a series of 118 patients, most of whom were type A (79%). The frequency of the other types was 5% for type B, 7% for type C, and 9% for the mixed type. The authors provided no immunohistochemistry findings.
Summary of the Findings From the 2 Case Series of Patients With Lymphomatoid Papulosis Published Since 2000.
| El Shabrawi-Caelen et al3 (n = 78) | Bennek et al2 2000 (n = 118) | |||||||
| A | B | C | Mixed | A | B | C | Mixed | |
| % | 75% | 4% | 13% | 8% | 79% | 5% | 7% | 9% |
| Wedge-shaped | 90% | 50% | 100% | – | – | – | – | – |
| CD30+ | 100% | 75% | 100% | – | – | – | – | – |
| CD56+ | 40% | 50% | 50% | – | – | – | – | – |
| CALCL | 0% | 0% | 0% | 0% | 10 (9%) | |||
| Hodgkin lymphoma | 3% | 0% | 9% | 14% | 2 (1.7%) | |||
| Mycosis fungoides | 3% | 0% | 9% | 9% | 11 (9.3%) | |||
| Papules | – | – | – | – | 68% | |||
| Treatment | – | – | – | – | ||||
| None | 56% | |||||||
| Psoralen–UV-A | 35% | |||||||
Abbreviation: CALCL, cutaneous anaplastic large-cell lymphoma.
It is important to note that it is not possible to differentiate LyP from other CD30+ lymphoproliferative disorders on immunohistochemistry findings. Although positivity demonstrates the presence of the disease, some cases are negative. The reasons why LyP is CD30– are not clear and have been reported mainly for type B disease.12 One possible explanation is that manifestations assumed to be compatible with LyP were really those subsequently reported for papular mycosis fungoides and, therefore, indicative of a CD30– disorder.13
CD3 is a pan–T-cell marker and, as such, positive in almost all cases of LyP. In our series, we found that it was more intensely positive in cells of the infiltrate accompanying large cells, thus suggesting the existence of 2 T-cell populations. However, as this finding was not described in the literature reviewed, further studies are warranted to confirm it.
We analyzed CD56, because 50% of the LyP biopsy specimens in the series by El Shabrawi-Caelen et al3 were positive for this marker. In our study, however, we found only 1 case of doubtful CD56 positivity. The literature review showed that positivity for CD56 in LyP remains open to debate and generally tends to be described as very rare.14 The reasons for such a discordant finding between 2 series published in the last 6 years remain unclear, although one explanation could be the different dilutions of reagent used: 1:20 in the study by El Shabrawi-Caelen et al in 2004, 1:40 in 3 cases of CD56+ LyP in the study by Flann et al14, and 1:25 in our series.
T-cell receptor testing in patients with LyP is useful for differentiating between lesions involving clonal rearrangement and those of a reactive nature. However, positivity or negativity of the T-cell receptor is merely orientative in clinical practice. Moreover, the role of clonal rearrangement in LyP remains unclear: in 2003, Gellrich et al9 reported clonality in small CD30– cells in the accompanying infiltrate, whereas in 2002, Steinhoff et al7 reported clonality in large CD30+ cells.
Classification of LyP into 3 histologic patterns, both in our study and in the literature reviewed,2,3 is more a convention than of relevance to symptoms, prognosis, and response to treatment. No apparent association exists between the histologic pattern and the variables studied.
Symptoms are self-limiting in most cases (approximately 60% to 70% according to the literature). However, the risk of progression from LyP to mycosis fungoides has been reported in 10% to 12% of cases, an association with Hodgkin lymphoma in 9% of cases, and an association with cutaneous anaplastic large-cell lymphoma in 9% of cases (Table 3). We detected progression to mycosis fungoides in only 12% of our patients; neither of the other associations was observed. A diagnosis of LyP after a diagnosis of mycosis fungoides must be made with caution, since it may reveal a transformation to large cell disease, thus necessitating a radical change in the treatment and prognosis of the initial mycosis fungoides. Knowledge of these associations in patients with LyP requires us to perform periodic follow-up and 6-monthly monitoring with laboratory tests (including lactate dehydrogenase) and chest x-ray.5
Most cases of LyP present with papular lesions, are self-limiting, and do not require treatment. More persistent outbreaks usually respond well to psoralen–UV-A, as was the case in 35% of the patients in the series by Bennek et al2 in 2000 and in 22% of our patients. Low-dose methotrexate is an effective alternative. In the very rare refractory cases, bexarotene,15 interferon alfa,16 topical 5-fluorouracil,17 and photodynamic therapy18 have been tried, with varying success.
We conclude that LyP affects middle-aged patients in the form of outbreaks of papules on the extremities and trunk. Most cases resolve spontaneously, and the response to treatment is good. The typical histologic finding is a wedge-shaped infiltrate of large CD30+ cells and, rarely, CD56+ cells. Follow-up is necessary, as the disease has been reported to be associated with other lymphoproliferative disorders.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article: Fernández-Guarino M, et al. Papulosis linfomatoide: hallazgos clínico-patológicos en 18 pacientes. Actas Dermosifiliogr.2012;103:388-93.


