



Los linfomas cutáneos primarios de células T distintos de la micosis fungoide, el síndrome de Sézary y los procesos linfoproliferativos cutáneos positivos para CD30 son poco frecuentes, representan menos del 5% de todos los linfomas cutáneos; generalmente se caracterizan por un fenotipo citotóxico y habitualmente presentan un comportamiento clínico agresivo. A menudo, los pacientes presentan o desarrollan enfermedad extracutánea poco después del diagnóstico. El manejo comúnmente incluye un enfoque multidisciplinario, se debe considerar un tratamiento sistémico intensivo y un trasplante de médula ósea. Los linfomas cutáneos primarios de células B representan aproximadamente el 30% de los linfomas cutáneos primarios. Incluyen un grupo heterogéneo de entidades con diferentes características clinicopatológicas y evolutivas. Suelen presentarse como pápulas, nódulos o tumores de coloración variable (rojo-violeta), solitarios o múltiples, que aparecen ocasionalmente agrupados o como lesiones generalizadas multifocales en el tronco, la cabeza o las extremidades. Se pueden distinguir 3grupos bien definidos: el linfoma cutáneo primario de células del centro del folicular y el linfoma cutáneo primario de células de la zona marginal, que siguen un curso clínico indolente, y el linfoma difuso cutáneo primario de células B grandes del tipo de las piernas, de curso agresivo.
Primary cutaneous T-cell lymphomas other than mycosis fungoides, Sézary syndrome, and lymphoproliferative CD30+ disorders are few, accounting for less than 5% of all cutaneous lymphomas. A cytotoxic phenotype is characteristic of these tumors, and their clinical behavior is usually aggressive. Patients often present with extracutaneous symptoms or develop them shortly after diagnosis. Management is usually multidisciplinary, and intensive systemic therapy and bone marrow transplantation should be considered. Cutaneous B-cell lymphomas account for approximately 30% of primary cutaneous lymphomas. They make up a heterogeneous group of tumors that have different clinical and pathological features. Clinical course also varies. Presenting as papules, nodules, or tumors of variable reddish–violaceous coloring, the lesions may be solitary or multiple and occasionally form clusters. There may also be generalized lesions, present at multiple sites on the trunk, head, or extremities. Three well-defined groups of primary cutaneous lymphoma have been reported: follicle center lymphoma; marginal zone lymphoma, which follows an indolent course; and a diffuse large B-cell lymphoma, leg type, which follows an aggressive course.
Los linfomas cutáneos primarios de células T distintos de la micosis fungoide, el síndrome de Sézary y los procesos linfoproliferativos cutáneos positivos para CD30 son poco frecuentes, representan menos del 5% de todos los linfomas cutáneos, generalmente se caracterizan por un fenotipo citotóxico y habitualmente presentan un comportamiento clínico agresivo. A menudo, los pacientes presentan o desarrollan enfermedad extracutánea poco después del diagnóstico. Su manejo implica una aproximación multidisciplinaria y puede plantear un tratamiento sistémico intensivo e incluso un trasplante de progenitores hematopoyéticos.
Linfoma de células T subcutáneo tipo paniculíticoEl linfoma de células T subcutáneo tipo paniculítico (LCCT-P) es un subtipo poco frecuente caracterizado clínicamente por lesiones similares a una paniculitis cuyas células neoplásicas corresponden a células T CD8+/CD4–/CD56–/TCR-αβ de fenotipo citotóxico (TIA-1, granzyme B, perforina)1. Los pacientes suelen referir lesiones nodulares inflamatorias cutáneas recurrentes de evolución indolente durante meses o años (con una supervivencia superior al 80% a los 5 años). La diseminación extracutánea es poco frecuente, aunque excepcionalmente puede asociarse con procesos autoinmunes (particularmente a lupus eritematoso)2. Recientemente, se ha descrito la presencia de mutaciones en HAVCR2/TIM3 en un grupo de pacientes con LCCT-P que podría asociarse con un síndrome hemofagocítico (pancitopenia, fiebre y hepatoesplenomegalia)3. Histológicamente, se observa un denso infiltrado linfoide localizado casi exclusivamente en el tejido celular subcutáneo. El infiltrado afecta tanto a los septos como a los lobulillos, con una distribución marginal característica de las células malignas atípicas en torno a los adipocitos con fenómenos de fagocitosis de detritus nucleares y hematíes (hemofagocitosis). No se asocia con una infección por virus de Epstein-Barr (VEB). El diagnóstico diferencial debe plantearse con otros LCCT con afectación predominante del tejido celular subcutáneo, especialmente el LCCT-γδ, o de formas cutáneas secundarias de linfomas sistémicos de células T. El tratamiento habitual son los corticoides orales a bajas dosis, metotrexato, interferón o incluso radioterapia en formas localizadas1.
Linfoma cutáneo de células T fenotipo γδEl LCCT-γδ es una neoplasia linfoide de evolución habitualmente agresiva. La presentación clínica suele ser heterogénea en forma de nódulos o placas con tendencia a la ulceración, solitarias o múltiples y de localización variable. Con frecuencia se desarrolla una afectación extracutánea característicamente extranodal (pulmón, sistema nervioso central, intestino, etc.) o un síndrome hemofagocítico4,5. Histopatológicamente, se evidencia una proliferación linfoide (nodular o difusa) formada por células pleomórficas en unas ocasiones de predominio epidermotropo y en otras en forma de infiltración intensa del tejido celular subcutáneo simulando a un LCCT-P. Pueden observarse histiocitos con un fenómeno de hemofagocitosis. Las células neoplásicas no suelen expresar ni el antígeno CD4 ni el CD8, son TCR-γδ/CD2+/CD3+/CD56+/VEB–6. El manejo del LCCT-γδ y de los linfomas cutáneos citotóxicos en general, en pacientes de edad avanzada o con comorbilidades asociadas, no contempla tratamientos agresivos, mientras que en otros casos el abordaje debería incluir la posibilidad de un trasplante alogénico de precursores hematopoyéticos. El papel de las mutaciones en el gen SETD2 recientemente descritas puede plantear en un futuro nuevas opciones de terapias dirigidas7.
Linfoma extranodal de células NK/T, tipo nasalEl linfoma de células natural killer (NK) o linfoma extranodal NK/T tipo nasal tiene una etiología claramente relacionada con una infección por el VEB. Este grupo de linfomas es más prevalente en zonas geográficas del Sudeste asiático, Centroamérica y Sudamérica8. La localización cutánea es la segunda en frecuencia después de la cavidad nasal. Es un linfoma de evolución clínica agresiva con una diseminación rápida a órganos extranodales (piel, tubo digestivo, etc.) y una supervivencia de un 20% a los 5 años9. Clínicamente, las lesiones suelen ser placas o nódulos eritematosos o violáceos múltiples con tendencia a la ulceración y, en los casos de localización nasal, una lesión nodular centrofacial destructiva (cavidad oral y tracto respiratorio superior) con clínica de obstrucción nasal, epistaxis, edema/inflamación de cabeza y cuello (granuloma letal de la línea media)8. La histología muestra un denso infiltrado dermo-hipodérmico con frecuente fenómeno de angiodesducción y extensas áreas de necrosis formados por células CD56+/CD2–/CD3ɛ+ citoplasmático, de fenotipo citotóxico (TIA-1+).
Proceso linfoproliferativo similar a hydroa vacciniformeLa nueva clasificación de la OMS-EORTC define el concepto de infección crónica por VEB que incluye las lesiones similares a hydroa vacciniforme (HV-like), que constituye un proceso linfoproliferativo T de fenotipo CD8+, con un antecedente frecuente de una respuesta exagerada a picaduras de mosquito y una expresión de marcadores de células NK. Se observa en algunas etnias o áreas geográficas específicas: América Latina y Asia. La HV-like suele observarse en niños o adultos jóvenes y se manifiesta en forma de pápulas y placas infiltradas, costras/ulceración, ampollas, edema, que se resuelven dejando cicatrices varioliformes localizadas en áreas expuestas. Presenta un riesgo de progresión a linfoma en la edad adulta. Puede asociarse con sintomatología sistémica: fiebre, pérdida peso, afectación del estado general, adenopatías, hepatoesplenomegalia, y anemia6,10.
Otros linfomas cutáneos T periféricos no específicosSe incluyen como linfomas T cutáneos no específicos aquellos procesos linfoproliferativos cutáneos de difícil inclusión en las categorías anteriores por sus características clínico-patológicas, inmunofenotípicas poco definidas. En general, corresponden a linfomas agresivos con morfología o expresión de marcadores citotóxicos y con variabilidad en la expresión de marcadores de célula T11.
Entidades provisionalesLinfoma cutáneo primario de células T CD8+ citotóxico epidermotropo agresivoEl linfoma cutáneo primario de células T CD8+ citotóxico epidermotropo agresivo (LCCTEA-CD8+) se manifiesta como máculas, placas y tumores generalizados a menudo ulcerados y lesiones hemorrágicas con frecuente afectación mucosa (oral, genital), aunque se han descrito lesiones indolentes de aspecto psoriasiforme o incluso similares a una micosis fungoide (MF). El LCCTEA-CD8+ desarrolla una evolución agresiva con una rápida diseminación extracutánea extranodal (pulmonar, sistema nervioso central, etc.) y muy mal pronóstico. Histológicamente, se observa una proliferación nodular o difusa de linfocitos atípicos con marcado epidermotropismo, necrosis y ulceración epidérmica con invasión y destrucción ocasional de los anejos cutáneos o estructuras vasculares. Las células neoplásicas corresponden a linfocitos de fenotipo citotóxico CD3+/CD7–/CD8+/CD45RA+ (CD2–/CD4–/CD5–/CD30–)/TIA-1+. El diagnóstico diferencial se plantea fundamentalmente con otros linfomas citotóxicos y con distintos procesos indolentes de fenotipo CD8+, como la reticulosis pagetoide, algunos casos de MF CD8+, papulosis linfomatoide tipo D o linfomas anaplásicos cutáneos CD30+12. Precisa de tratamiento sistémico intenso precoz, incluida la opción de trasplante alogénico de progenitores hematopoyéticos13.
Linfoma cutáneo primario acral de células CD8 positivasEl LCCT-CD8+ acral se presenta con lesiones nodulares solitarias o en distribución bilateral y simétrica, de crecimiento lento y evolución no agresiva, localizadas en los pabellones auriculares o en otras zonas acrales (nariz, dedos). La localización y las características de las lesiones han llevado a sugerir el posible carácter reactivo de esta entidad. Los hallazgos histopatológicos corresponden a un denso infiltrado difuso monomorfo no epidermotropo constituido por células T atípicas de tamaño pequeño-intermedio afectando difusamente a la dermis y el tejido celular subcutáneo. Las células tumorales expresan un inmunofenotipo citotóxico CD8+/TIA1+/granzima B–14. La expresión perinuclear puntiforme del antígeno CD68 sería un posible factor diferencial característico no presente en otros linfomas T citotóxicos agresivos.
Procesos linfoproliferativos de células T pleomórficas de pequeño/mediano tamaño CD4 positivasEl concepto de proceso linfoproliferativo de células T pleomórficas de pequeño/mediano tamaño CD4 positivas define casos de proliferaciones cutáneas T de evolución indolente que se presentan como nódulos solitarios, o en escaso número, agrupados, en el polo cefálico o tronco superior y cuya histología muestra un denso infiltrado linfoide dérmico de linfocitos T CD3+/CD4+/CD5+/CD7–/CD30– que asocian marcadores de células del centro folicular como BCL6+/CD10+/CXCL13/ICOS. El antígeno PD1, aunque no es específico, es el principal marcador necesario para el diagnóstico de esta entidad5,15,16. En la clasificación actual de la OMS de neoplasias hematológicas no se lo considera un linfoma verdadero, sino un proceso linfoproliferativo T muy probablemente de carácter reactivo.
Linfomas cutáneos primarios de células BLos linfomas cutáneos primarios de células B (LCCB) representan aproximadamente el 30% de los linfomas cutáneos primarios. Incluyen un grupo heterogéneo de entidades con unas características clínico-patológicas y evolutivas diferenciadas. Incluyen el LCCB del centro folicular (LCCB-CF), el LCCB de la zona marginal (LCCB-ZM) y el LCCB difuso de células grandes, tipo piernas (LCCB-DG)5,17.
El LCCB-ZM y LCCB-CF son procesos linfoproliferativos de comportamiento clínico indolente, mientras que el LCCB-DG tipo piernas representa una evolución agresiva y cuya aproximación terapéutica es similar a la de un linfoma de células B difuso sistémico18,19. El estudio de extensión inicial de un LCCB debe descartar una afectación cutánea de un linfoma sistémico (linfoma cutáneo secundario) (tabla 1)17,19. El diagnóstico diferencial entre el LCCB-ZM, el LCCB-CF, una hiperplasia linfoide reactiva (HLR) o un proceso linfoproliferativo de células T pleomórficas de pequeño/mediano tamaño CD4 positivas puede ocasionalmente plantear dificultades al solaparse aspectos clínicos e histológicos (población mixta de células T y B, citomorfología variable con predominio de células pequeñas)18.
Exploraciones complementarias para el diagnóstico y estadificación de los linfomas cutáneos de células B
| Hemograma completo con recuento y fórmula |
| Bioquímica convencional, determinación sérica de LDH y β-2-microglobulina, serología Borrelia burgdorferi |
| Estudio de clonalidad en sangre periférica, así como una citometría de flujo e inmunofenotipo de los linfocitos circulantes |
| TAC con contraste de tórax, abdomen y pelvis (importante realizar ventana pélvica o ECO testicular en los LCCB-DG para descartar linfoma testicular) y en caso de lesiones localizadas en el polo cefálico también una TAC cervical Alternativamente, puede realizarse un estudio de extensión mediante PET o TAC-PET |
| Biopsia ganglionar si adenopatía > 1cm o elevada actividad en PET |
| Biopsia de médula ósea no necesaria en LCCB-ZM, opcional/recomendable en LCCB-CF, siempre en LCCB-DG |
| Considerar un cribado de enfermedad autoinmune o afectación territorio MALT por linfoma según clínica asociada (en los LCCB-ZM), así como mutaciones MYD88 para diagnóstico diferencial proliferaciones linfoplasmacíticas |
| El sistema de estadificación TNM de los LCCB es similar al propuesto para los linfomas cutáneos de células T distintos de la micosis fungoide y el síndrome de Sézary41 |
Fuente: Senff et al.17.
El LCCB-CF es el subtipo más frecuente de linfoma cutáneo primario de células B (50-60% de los LCCB). Se manifiesta clínicamente en adultos en forma de pápulas eritematosas, placas o nódulos sin tendencia a ulcerarse, solitarios o múltiples, ocasionalmente agrupadas afectando a menudo el polo cefálico o el tronco. La afectación extracutánea puede llegar a observarse hasta en un 10% de los casos y suele ser un linfoma con un pronóstico favorable con una supervivencia a los 5 años superior al 90%17,20. No es infrecuente observar recurrencias limitadas a la piel.
Histológicamente, los LCCB-CF se caracterizan por un infiltrado linfoide dérmico con un patrón nodular folicular (formación de folículos linfoides expandidos y confluentes), difuso o mixto, respetando la dermis papilar y extendiéndose ocasionalmente al tejido celular subcutáneo. Se observan folículos linfoides irregulares, superpuestos, con centros germinales reconocibles constituidos por agregados de centrocitos y centroblastos sobre una red de células dendríticas bien estructurada, no presentan polaridad (zonas claras y oscuras) y poseen unos mantos bien formados. En las variantes con patrón difuso no se observa la formación de folículos linfoides20. Las células neoplásicas corresponden a linfocitos B (CD19+, CD20+, CD79a+, PAX-5+), que expresan marcadores de células del centro germinal (BCL-6, CD10) y, a diferencia de los linfomas centrofoliculares nodales, suelen ser BCL2–. No expresan marcadores de célula B activada (MUM1–/FOXP1–), lo que permite diferenciarlos de los LCCB-DG. La presencia de un índice de proliferación (Ki-67) no muy intenso (< 50%) en los centros germinales apoya el diagnóstico. Una expresión de BCL2 obligaría a excluir un linfoma folicular sistémico, un LCCB-ZM o un LCCB-DG21,22. No se han identificado alteraciones genéticas específicas. La translocación (14;18) (q32;q21)-IgH/BCL2 no suele detectarse, aunque en algunas series se ha detectado en un 10% de los casos. Ocasionalmente, se han descrito translocaciones entre los genes IgH y BCL6, amplificaciones en 2p16.31-REL o pérdidas en 14q32.32.23,24. El diagnóstico diferencial de algunos LCCB-CF debe establecerse con las HLR de células B, especialmente con un patrón folicular. Sin embargo, los folículos linfoides observados en las HLR suelen ser más monomorfos, están mejor formados que en los LCCB-CF con polaridad y no presentan una expansión de células del centro folicular hacia las zonas interfoliculares. Los estudios de clonalidad pueden ayudar a determinar el origen monoclonal o reactivo del proceso21.
Linfoma cutáneo primario de células B de la zona marginalEl LCCB-ZM no se reconoce como una entidad individualizada en la clasificación actual de la OMS de neoplasias hematológicas, sino se incluye dentro del grupo de los linfomas tipo MALT (mucosa associated lymphoid tissue), que se desarrollan en localizaciones extranodales o mucosas como el estómago, las glándulas salivales, órbita, tiroides, mama o pulmón5,18,19,25. Los linfomas tipo MALT parecen desarrollarse en tejidos en los que existe una activación linfoide persistente como consecuencia de una estimulación antigénica crónica, como Helicobacter pylori en los linfomas MALT gástricos, Campylobacer jejuni en los linfomas intestinales o una infección por Borrelia burgdorferi, vacunas o tatuajes en los cutáneos. Los LCCB-ZM se caracterizan clínicamente por la presencia de pápulas, placas o nódulos, solitarias o múltiples, en el tronco o las extremidades en individuos adultos. Las recurrencias exclusivamente cutáneas se observan en hasta un 50% de estos pacientes, aunque la afectación extracutánea es excepcional. Presenta una baja tasa de mortalidad (supervivencia 100% a los 5 años)17,25-27.
Histológicamente se caracteriza por una infiltración nodular, difusa, a menudo perianexial, en la dermis reticular y el tejido celular subcutáneo, respetando la dermis papilar y la epidermis formada por linfocitos de pequeño a mediano tamaño (monocitoides), con núcleo indentado y citoplasma claro, similares a las células de la zona marginal. Con frecuencia se observan escasas células de gran tamaño (similares a centroblastos) y un número variable de linfocitos de morfología plasmacitoide y células plasmáticas lo que facilita el diagnóstico. Se observan centros germinales reactivos y una expansión de las células de la zona marginal neoplásicas hacia las áreas interfoliculares17,21. Son linfocitos B maduros (CD20+, CD22+, CD79a), con una expresión aberrante de CD43+. Expresan BCL2+ y son CD10–/BCL6–. Con frecuencia, existe una población linfoide T acompañante prominente o predominante, lo que puede planear dificultades diagnósticas en diferenciar estas lesiones HLR o de un proceso linfoproliferativo T CD4+ pleomórfico de célula pequeña/intermedia. Se han descrito 2subgrupos de LCCB-ZM: un grupo poco frecuente con expresión de IgM/CXCR3, similar a otros linfomas tipo MALT, y un grupo con expresión de IgG+/IgG4/CXCR3– o «class-switched». Puede demostrarse una expresión monotípica de las cadenas ligeras de las inmunoglobulinas en una mayoría de casos28.
El estudio de clonalidad mediante técnicas de PCR permite asimismo detectar la presencia de una clona dominante en la mayoría de los casos. No sería necesario realizar una biopsia de médula ósea, debería descartarse una infección subyacente o una situación de estimulación antigénica crónica incluida posibles enfermedades autoinmunes, así como la presencia de proliferación linfoide en otros territorios MALT o mutaciones en MYD88 para el diagnóstico diferencial de otros procesos linfoplasmacíticos con presentación cutánea secundaria. La estimulación crónica de este tejido linfoide lo hace genéticamente inestable, facilitando la adquisición de alteraciones genéticas como la trisomía 3, trisomía 18, t(1;14) (p22;q32), t(11;18) (q21;q21), t(14;18) (q32;q21)-IgH/MALT, t(3;14) (q27;q32) o t(3;14) (p14.1;q32), lo que daría lugar a la transformación linfomatosa. Dichas alteraciones citogenéticas se detectan en un número limitado de LCCB-ZM (10-15%)29.
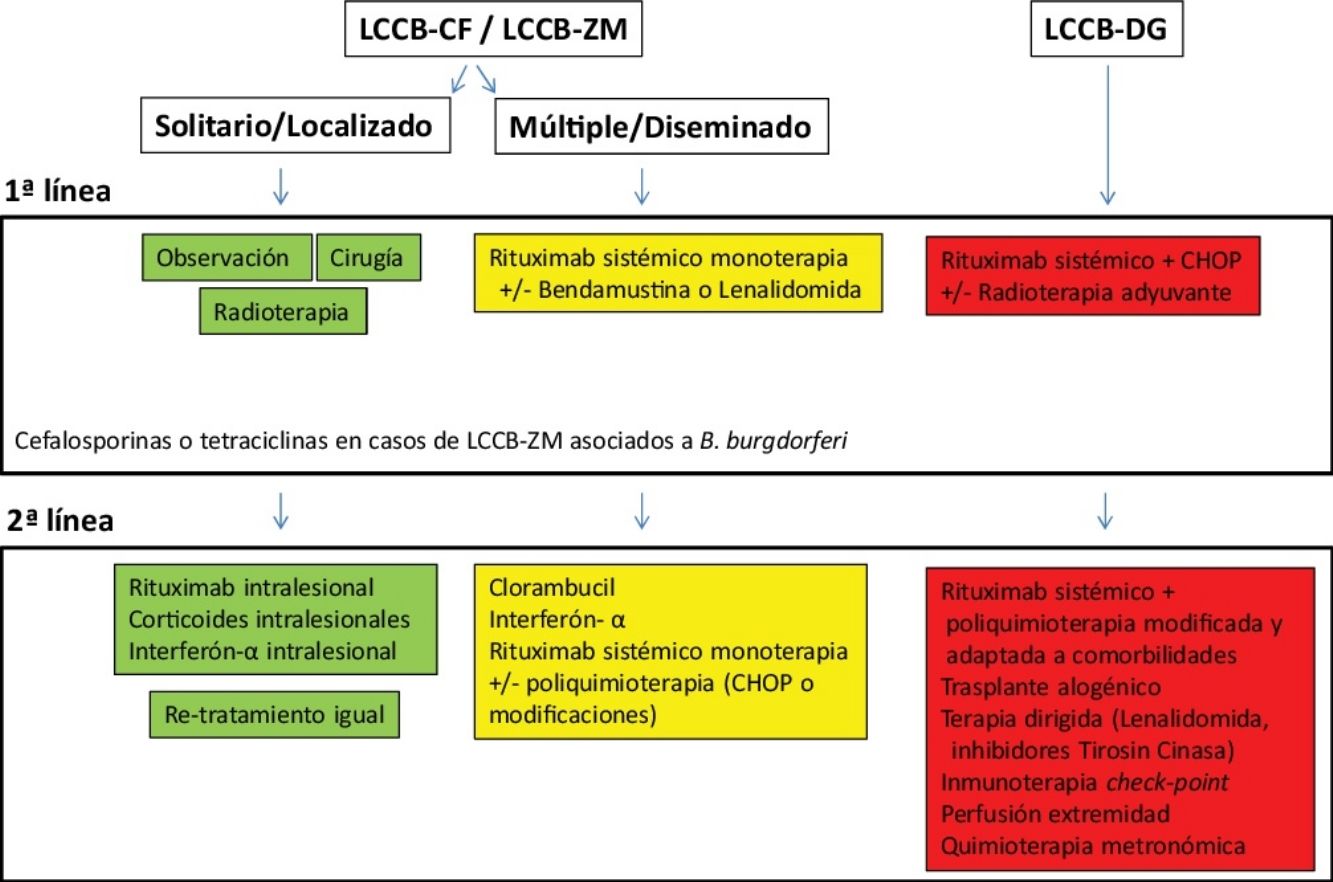
Tratamiento de los linfomas cutáneos de células B indolentesEl tratamiento de los LCCB indolentes (LCCB-CF y LCCB-ZM) es muy similar. Son linfomas altamente radiosensibles, por lo que la radioterapia local con electrones o fotones es una opción terapéutica para lesiones tumorales solitarias o agrupadas de abordaje quirúrgico complejo (fig. 1). Se recomienda unas dosis de 20 a 36Gy para los LCCB-ZM y 30Gy para LCCB-CF, aunque, recientemente, se han propuesto esquemas de tratamiento con dosis bajas de radiación para reducir la toxicidad30,31. La opción de rituximab intralesional ha demostrado ser eficaz y segura para aquellos casos en los que no son posibles otras opciones, evitando el desarrollo de cicatrices quirúrgicas o alopecia posradioterapia32. En pacientes con múltiples lesiones, suelen recomendarse tratamientos sistémicos (rituximab intravenoso en monoterapia o asociado a monoquimioterapia).
Linfoma cutáneo primario de células B difuso de células grandes, tipo piernas
El LCCB-DG es un proceso linfoproliferativo maligno linfoide de células grandes (centroblastos, inmunoblastos) con fenotipo de célula B activada. Suele observarse en individuos de edad avanzada y presenta una supervivencia a los 5 años de un 50%5,18-20. El LCCB-DG tipo piernas suele manifestarse con lesiones nodulares o tumores solitarios o agrupados, con tendencia a la ulceración. Aunque se describió inicialmente en una o ambas piernas, puede desarrollarse en otras localizaciones5,18-20,33,34. El estudio histopatológico muestra un infiltrado difuso monomorfo que suele ocupar toda la dermis, afectando ocasionalmente al tejido celular subcutáneo formado por células redondas de gran tamaño BCL-2+/MUM-1+/FOXP1+/BCL6+. Suelen expresar MYC, IgM, y P635,18-20,22,33. El perfil genético del LCCB-DG es similar al observado en los linfomas difusos de células B grandes sistémicos, con mutaciones en genes implicados en la vía de señalización NF-κB (CD79B, PM1CARD11 y MYD88 en 60-75% de los casos), remodelación del ADN (TBL1XR1, CREBBP, IRF4 y HIST1H1E), translocaciones o amplificaciones en los genes IgH, BCL6, MYC o FOXP1, así como deleciones o hipermetilación del promotor de p1630,33,35,36.
Los LCCB-DG se tratan de forma similar a los linfomas difusos sistémicos con rituximab asociado a poliquimoterapia (CHOP) o pautas adaptadas a las comorbilidades de los pacientes. La radioterapia local se utiliza como tratamiento adyuvante o paliativo. El conocimiento exhaustivo de vías moleculares específicas en estos linfomas agresivos plantea posibilidades de terapias dirigidas. Por otro lado, existen desarrollos con inmunoterapia con inhibidores check-point y la posibilidad de trasplante alogénico precursores hematopoyéticos en casos seleccionados5,17,19,20.
Linfoma B de células grandes intravascularEl linfoma B de células grandes intravascular (LCCB-DGIV) es un proceso poco frecuente de evolución agresiva que posee como característica particular una infiltración linfoide maligna de células B activadas MUM-1+/BCL2+ en el interior de las luces vasculares37. El LCCB-DGIV tiende a afectar a distintos órganos y específicamente a la piel (máculas, telangiectasias) y el sistema nervioso central. En un número significativo de casos, las lesiones cutáneas son la primera manifestación de este proceso, aunque la mayoría de los pacientes presentan una enfermedad sistémica en el momento del diagnóstico37.
Entidades provisionalesÚlcera mucocutánea asociada a infección por el virus de Epstein-BarrLa úlcera mucocutánea secundaria a VEB se ha incluido como una entidad provisional dentro de la clasificación de neoplasias linfoides. Se observa en individuos de edad avanzada o tratados mediante una situación de inmunosupresión. Clínicamente, suele manifestarse como una o varias úlceras de bordes bien definidos en la mucosa orofaríngea o en el tracto gastrointestinal. Ocasionalmente pueden asociar también lesiones cutáneas. Suele ser un proceso autolimitado, que mejora tras la resolución de la inmunosupresión5,23,38. Histológicamente, se caracteriza por una ulceración epitelial, con hiperplasia seudoepiteliomatosa y por un infiltrado dérmico/submucoso polimorfo con linfocitos, inmunoblastos, células similares a células de Reed-Sternberg, células linfoides de pequeño tamaño, células plasmáticas y eosinófilos, junto con extensas áreas de necrosis. Las células atípicas de aspecto blástico corresponden a linfocitos B que expresan PAX-5+/OCT-2+/EBER+/CD30+/CD15+38.
Otros procesos hematológicos no linfoproliferativos con afectación cutánea predominante o característicaLeucemia de células dendríticas (neoplasia hematodérmica CD4/CD56)Es una neoplasia hematológica maligna agresiva de células dendríticas precursoras plasmacitoides o mieloides en la que con frecuencia las lesiones cutáneas son la primera manifestación de la enfermedad, aunque la afectación multisistémica (sangre periférica, médula ósea) suele hallarse presente en el momento del diagnóstico o hacerse evidente poco después del mismo23,39. Las lesiones cutáneas habitualmente corresponden a nódulos solitarios o múltiples característicamente de tonalidad purpúrica de crecimiento rápido localizados en tronco y polo cefálico, sin tendencia a la ulceración. Histopatológicamente, se observa una infiltración dérmica difusa formada por una población celular monomorfa no epidermotropa con una citomorfología similar a linfoblastos o mieloblastos con expresión de CD123/TCL1 y fenotipo CD4+/CD56+/CD8–/CD7±/CD2±/CD45RA+/CD3–23,39. La quimioterapia agresiva consigue remisiones completas, si bien las recaídas suelen ser precoces y la supervivencia global no suele ser superior a un año, por lo que puede plantearse como tratamiento el trasplante de precursores hematopoyéticos23. Recientemente, se ha aprobado, por la Food and Drug Administration, tagraxofusp, proteína de fusión de la toxina diftérica con interleucina-3, como posible tratramiento de esta entidad40.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



