


There are 3 types of leiomyosarcoma of the skin: dermal, subcutaneous, and metastatic cutaneous. Dermal leiomyosarcoma arises from smooth muscle fibers in arrector pili muscles, genital dartos muscles, and the nipple-areola complex. It is an intermediate-grade tumor associated with a tendency for local recurrence (24%) and low metastatic potential (4%). Subcutaneous leiomyosarcoma originates from smooth muscle in blood vessel walls and has higher rates of local recurrence (37%) and metastasis (43%).
Plemorphic dermal sarcoma typically affects elderly patients and arises in sun-exposed areas (e.g., the scalp). Its histologic and immunohistochemical characteristics are similar to those of atypical fibroxanthoma, but it is more aggressive (metastasis rate of 10%-20%). Histologically, it can be distinguished from atypical fibroxanthoma by the observation of subcutaneous tissue invasion, perineural invasion, and foci of necrosis.
El leiomiosarcoma de la piel se clasifica en tres grupos: dérmico, hipodérmico y cutáneo metastásico. El dérmico se origina de las fibras musculares lisas del músculo erector del pelo, dartos genital o de la areola mamaria. Se considera un tumor de malignidad intermedia, con tendencia a la recidiva local (24%) y un bajo riesgo de metástasis (4%). El leiomiosarcoma hipodérmico se origina de las paredes musculares de los vasos, y se caracteriza por presentar una mayor tasa de recidiva local (37%) y metástasis (43%).
El sarcoma pelomórfico dérmico aparece habitualmente en pacientes ancianos y se localiza característicamente en zonas de piel fotoexpuesta (cuero cabelludo). Comparte características histológicas e inmunohistoquímicas con el fibroxantoma atípico, pero con un comportamiento más agresivo (metástasis en el 10-20%). Los criterios histológicos que lo diferencian son la infiltración del tejido celular subcutáneo, la infiltración perineural y la presencia de focos de necrosis.
Leiomyosarcoma (LMS) is a tumor derived from smooth muscle that arises in deep soft tissue, the uterus, and, more rarely, the dermis. It accounts for approximately 5% to 10% of all sarcomas, but just 2% to 3% of cutaneous sarcomas (0.04% of all skin tumors).1,2 It is the third most common cutaneous sarcoma, after dermatofibrosarcoma and Kaposi sarcoma. LMSs located in the retroperitoneum and abdominal cavity form the most common subgroup of LMS and are much more aggressive than other variants.
LMS of the skin has traditionally been classified into 3 major groups, each with different prognostic implications: cutaneous (dermal) LMS, subcutaneous LMS, and metastatic LMS.3 The deeper the lesion, the worse the prognosis. Cutaneous LMS arises from the smooth muscle fibers of arrector pili muscle, the genital dartos muscle, or the nipple-areola complex, while subcutaneous LMS originates from smooth muscle in blood vessel walls.4 Both entities cause metastases that can affect the skin. Most cutaneous metastases, however, are from retroperitoneal LMS.
Cutaneous LMS is a tumor of intermediate malignancy that has a tendency to recur locally (24%) and low metastatic potential (4%).5 In view of this, Kraft et al.6 proposed that cutaneous LMSs with minimal subcutaneous involvement should not be considered sarcomas and suggested instead naming them atypical intradermal smooth muscle neoplasms. This term, however, has not had much uptake. Dermatofibrosarcoma protuberans, for instance, has a much lower tendency to metastasize to the skin, yet is still considered a sarcoma. Subcutaneous LMS is characterized by higher rates of local recurrence (37%) and distant metastasis (43%). Finally, cutaneous metastases from LMS indicate progression of a primary tumor, generally of visceral origin, and are associated with an approximate survival of 16 months from the time of detection.7
Before a definitive diagnosis of a primary LMS of the skin can be established, it is essential to rule out metastasis from an LMS in the deep tissues or organs, particularly if the tumor is subcutaneous.
Clinical CharacteristicsLMS can occur at any age, but it mostly affects older adults, with a peak incidence between 50 and 70 years. It is more common in men (male to female ratio, 3:1) and appears to be more common in whites.3 Most LMSs involving the skin develop de novo and are unrelated to a previous piloleiomyoma-like lesion. A mutation in the fumarate hydratase gene was recently discovered in patients with hereditary leiomyomatosis and renal cell cancer syndrome.8 LMS has also been described in areas previously treated with radiation therapy, and some patients have reported a history of trauma or scarring in the area. Most patients, however, have no known triggers.
Fifty percent of cutaneous LMSs are located on the extensor surface of the lower limbs, and less frequently on the scalp and face,9 although there have been reports of tumors involving the trunk, lip, genital region (scrotum, vulva, and penis), and buttocks.
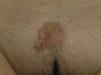
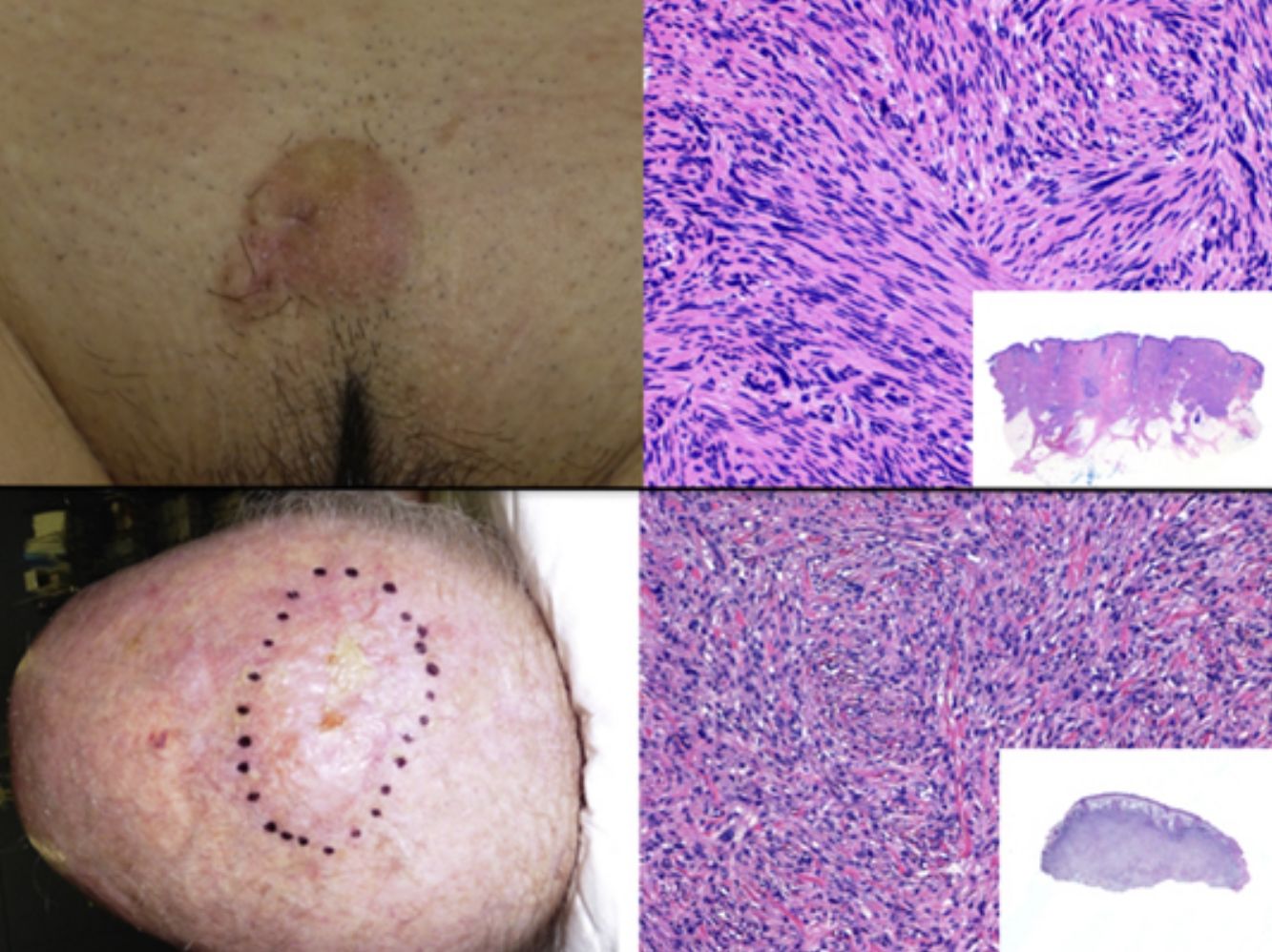
The clinical presentation of LMS is nonspecific. The most common presentation is a firm, solitary nodule with a smooth, pinkish surface, or a more exophytic tumor that has a reddish or brown color (Fig. 1). Clinically, subcutaneous lesions appear to be better circumscribed than their cutaneous counterparts, and they are reminiscent of lipomas, but with a more solid consistency. There have been reports of plaque-like LMS with multiple nodules that form clusters and are very indurated on palpation. Cutaneous lesions typically grow slowly and are larger than subcutaneous lesions, with a size of between 1 and 3.5cm (range, 0.5-19cm). They are frequently painful on palpation (63%). Spontaneous pain is also reported but is less common (25%).9 Patients may experience pruritus, a burning sensation, and paresthesia.3,9
Histopathologic Characteristics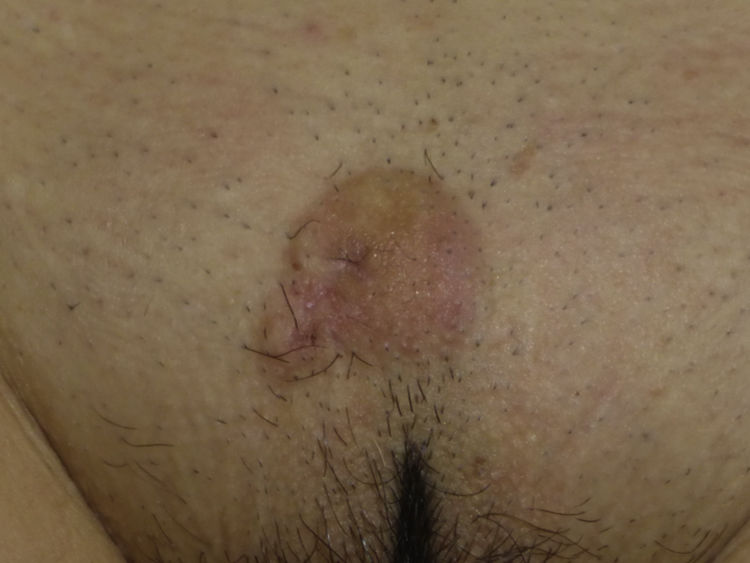
The biopsy specimen must include subcutaneous tissue.
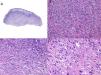
Histologic examination of cutaneous LMS generally reveals a poorly circumscribed lesion occupying the full thickness of the dermis and occasionally extending into the subcutaneous tissue (Fig. 2A and B). Subcutaneous LMSs are better circumscribed; they compress the adjacent tissue and are located entirely in the subcutaneous layer, with sparing of the dermis. In both cases, low magnification shows interlacing fascicles of smooth muscle fibers. The cells are spindle-shaped and have elongated nuclei with blunt ends, an unremarkable nucleolus, and eosinophilic fibrillar cytoplasm (Fig. 2C and D). Several cells have a clear, perinuclear halo characteristically seen in muscle cells.4,10,11
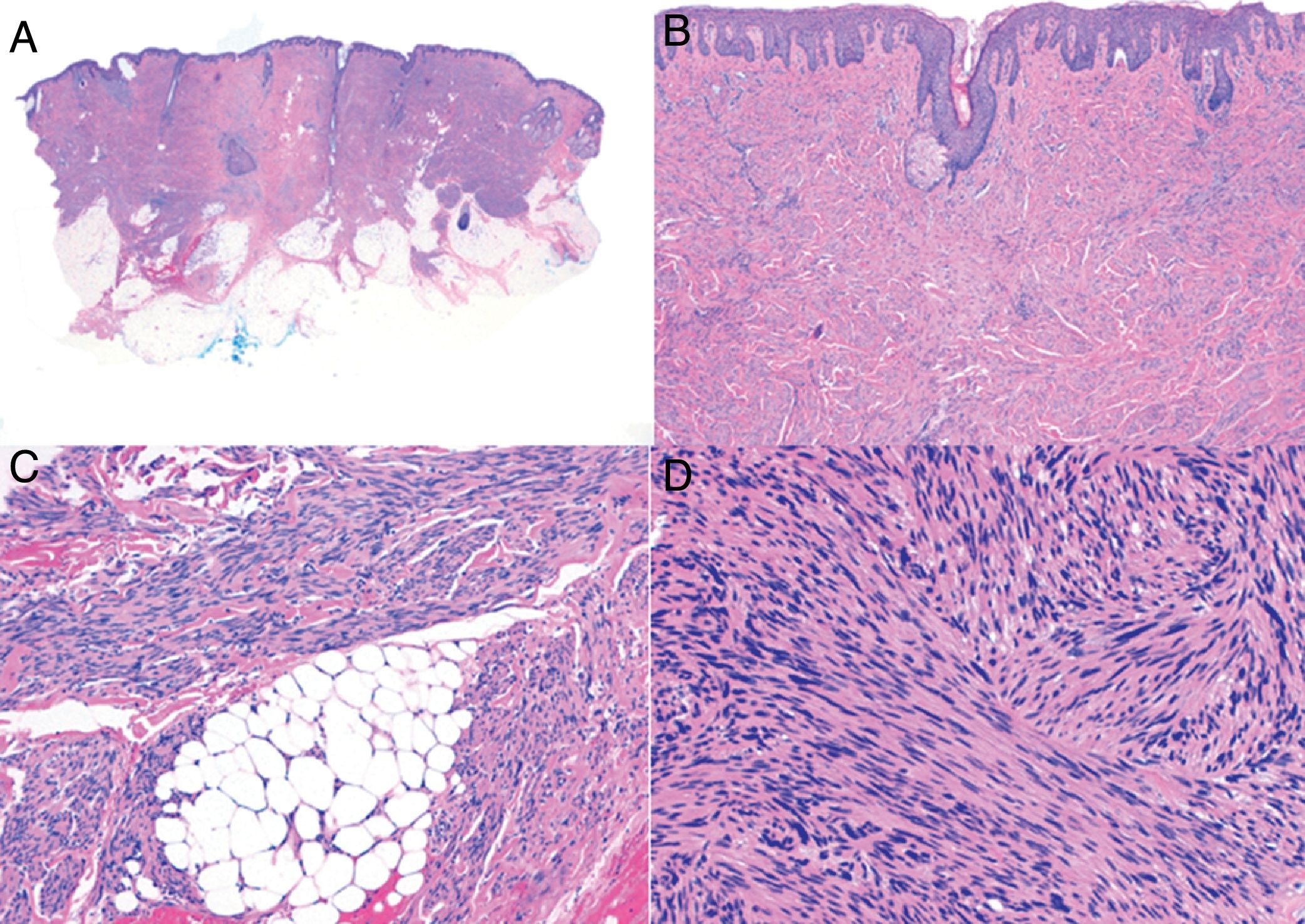
Characteristic histologic features of leiomyosarcoma. A, Panoramic view of a poorly circumscribed dermal tumor invading the subcutaneous tissue. B, Higher-magnification view showing interlacing fascicles of nonuniformly arranged spindle cells in the dermis reminiscent of muscle fibers. C, Invasion of subcutaneous tissue. D, Fascicles of pleomorphic spindle cells intersecting each other at a right angle; mitotic figures.
Two histologic growth patterns have been described: nodular and diffuse.12 The nodular pattern is characterized by greater cellularity, atypia, and a higher number of mitotic figures, while the diffuse pattern is characterized by less cellularity and pleomorphism and fewer mitotic figures.13
Cutaneous LMS with a diffuse growth pattern has little cellular atypia and can therefore be difficult to distinguish from leiomyoma. Although a more invasive pattern and greater cellularity point to the malignant variant, observation of mitosis is key for confirmation.
Several histopathologic variants of LMS have been described, including epithelioid LMS,14 LMS with multinucleated giant cells,15 granular cell LMS,16 and sclerotic LMS.17 Considerable desmoplasia has also been described and can complicate diagnosis. There have also been reports of myxoid and pleomorphic variants of subcutaneous LMS.4
When there is histologic evidence of LMS, an immunohistochemical study must be performed to rule out spindle cell tumors with similar histologic features. Well-differentiated LMSs show positive staining for vimentin, desmin, h-caldesmon, muscle specific actin, α-smooth muscle actin, and smooth muscle myosin.4 More poorly differentiated and subcutaneous lesions often test negative for desmin.18,19 At least 2 smooth muscle markers must be included in the panel to confirm a diagnosis of LMS. Protein S-100 is sometimes positive, as are cytokeratins. Other immunohistochemical stains used to rule out other spindle cell tumors (spindle cell carcinoma, desmoplastic melanoma, dermatofibrosarcoma protuberans, malignant peripheral nerve sheath tumors, and vascular tumors) are EMA, CD34, CD117, CEA, HMB45, Mart-1, Melan A, and CK7. These are all negative in LMS.
StagingThere is no specific TNM staging system for LMS and tumors are therefore staged using the classification system for soft tissue sarcomas in the American Joint Committee on Cancer Cancer Staging Manual.
Prognosis in LMS varies depending on whether the lesion is cutaneous or subcutaneous. Cutaneous LMS has a tendency to recur locally (24%), but rarely metastasizes (4%), while subcutaneous LMS is more like to recur locally (37%) and metastasize (43%). The respective staging and follow-up approaches are therefore different.
Cutaneous LMS
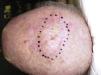
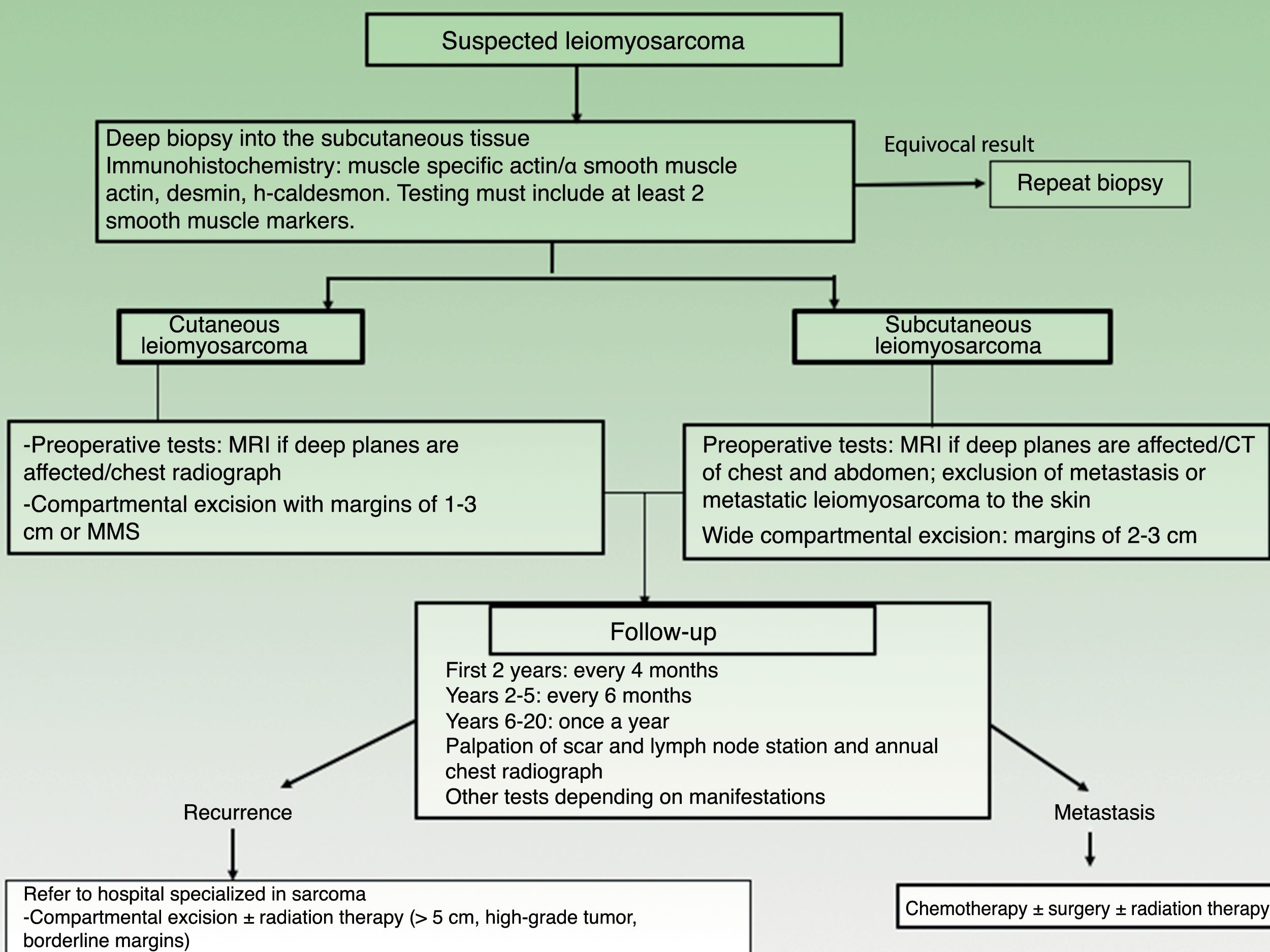
Magnetic resonance imaging (MRI) of the area surrounding a cutaneous LMS is recommended prior to surgery, especially for large, indurated, or difficult-to-access lesions (eg, on the head). Ultrasound may well be a useful aid for dermatologists and could even replace MRI (Fig. 3), but no studies have yet compared the 2 options. A chest radiograph should be also performed before surgery.
Subcutaneous LMS
An MRI of the area and computed tomography (CT) scan of the chest and abdomen should always be performed before operating on a subcutaneous LMS to rule out metastasis from a tumor in the deeper tissues20 (Fig. 3).
Treatment and PrognosisConsidering the low incidence of cutaneous LMS, patients should always be referred for treatment at a hospital specialized in sarcoma.
Complete surgical excision is the treatment of choice for LMS. The best results are obtained with wide local excision or Mohs micrographic surgery (MMS), which offers a more precise analysis of margins.
The main surgical dilemma in LMS is margin width, as there are no clear guidelines on recommended widths in the literature. Wide local excision with margins of 3cm to 5cm used to be the recommended approach, but nowadays, similar results are achieved using more conservative margins of between 1cm and 3cm30,21–24 (Fig. 3). Deep margins should include the fascia and, in the case of more invasive tumors, the muscle.
There is very little experience with the use of MMS in cutaneous LMS. The literature contains reports on approximately 50 cases, and the recurrence rates described range between 0% and 13%.3,22,23,25 These rates are much lower than those observed for conventional surgery, which is associated with recurrence rates of between 9% and 40%.3,21–23 MMS would therefore appear to be a good surgical option for superficial cutaneous LMS.
The role of radiation therapy in cutaneous LMS is still a topic of debate. Adjuvant radiation therapy may be of particular value in patients with positive or borderline surgical margins or who are not candidates for a second operation; this is particularly the case for patients with deep-seated or high-grade tumors. Factors associated with a worse prognosis are a subcutaneous or acral location, a size larger than 5cm, aneuploidy, and vascular invasion.13 Radiation therapy can also be used for local palliative control in patients with metastasis.
Chemotherapy is essential in patients with metastatic disease. The most widely used agents are doxorubicin and ifosfamide, gemcitabine and taxotere, dacarbazine, and trabectedin. Chemotherapy is not curative, but it can delay disease progression.26
Follow-upThere are no standardized follow-up guidelines for LMS, but a clinical check-up every 4 months is recommended for the first 2 years to aid the early detection of local recurrences. After that, 6-monthly visits are recommended for the following 3 years (up to year 5 after surgery) and then once a year up to year 20, as very late recurrences have been described.3,11 Standardized guidelines on radiologic studies for postoperative follow-up are also lacking for cutaneous sarcoma (Fig. 3). Nevertheless, a simple annual chest radiograph for the first 5 years after surgery and clinical evaluation of the surgical bed and locoregional lymph nodes would appear to be a good strategy.26 MRI can occasionally be of value, particularly in the case of recurrent or subcutaneous lesions or complicated surgery.
The metastasis rates for cutaneous and subcutaneous LMS are 4% (range, 3%-14%) and 43% (range, 21%-62%), respectively.3 These tumors primarily spread through the bloodstream and affect the lungs, skin, and less frequently, the regional lymph nodes. The ideal staging test for patients with confirmed or suspected disseminated disease is a spiral CT scan of the chest and abdomen.26
Pleomorphic Dermal SarcomaIntroductionPleomorphic undifferentiated sarcoma, formerly known as malignant fibrous histiocytoma, is a malignant soft-tissue tumor (sarcoma) that is typically highly pleomorphic and has no characteristic immunohistochemical features that point to a specific line of differentiation.27–32
Pleomorphic undifferentiated sarcoma that originates in the skin is known as pleomorphic dermal sarcoma (PDS). It has very similar histologic features to atypical fibroxanthoma, but has a worse prognosis.33,34 Because of the confusion generated by the different names classically given to these tumors and the update published by the World Health Organization in 2013,31 there are very few large series of PDS to add to the body of knowledge and from which to draw conclusions on adequate management.
DefinitionPDS is a cutaneous tumor of uncertain histogenesis. Its clinical characteristics are largely nonspecific and it is histologically and immunohistochemically very similar to atypical fibroxanthoma, although its behavior is more aggressive.29
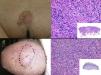
Clinical PresentationPDS typically affects elderly patients and is located in areas of sun-exposed skin, typically the head (Fig. 4) and in particular the scalp. As indicated by Tardío et al.,33 other tumors must be ruled out and a diagnosis of PDS should be treated with suspicion if the tumor does not involve sun-damaged skin in an elderly patient. PDS manifests as an exophytic, asymmetric tumor that is fast growing and often ulcerated and hemorrhagic. Mean tumor size is 2.2cm to 2.5cm,33,34 although sizes ranging from just a few millimeters to several centimeters have been reported. Clinically, the tumor is indistinguishable from atypical fibroxanthoma, but it has a larger subclinical extension. It does not therefore typically raise clinical suspicion and tends to be diagnosed as squamous cell carcinoma.
Histopathologic Characteristics and Diagnosis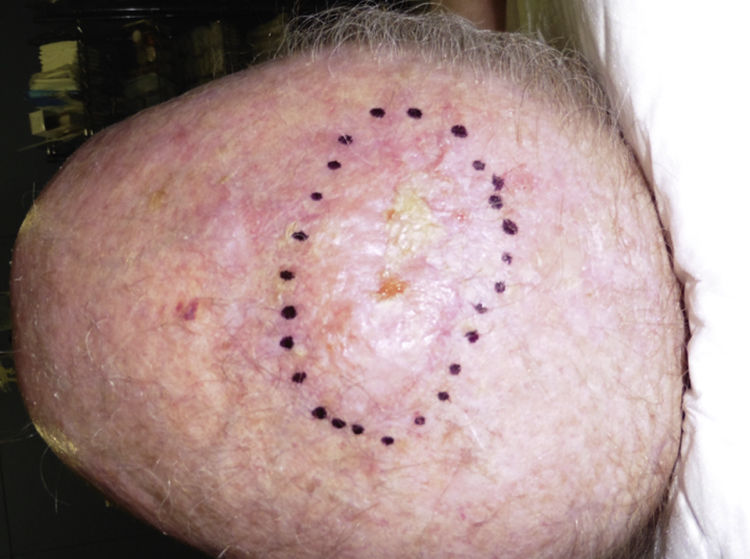
As PDS is of unknown origin and histogenesis, the diagnosis is one of exclusion.
PDS is confined to the dermis; there is no connection to the epidermis or Grenz zone (area of normal collagen in the dermis that separates the epidermis and the tumor).33,34 The tumor is formed by 2 populations of cells present in different proportions: atypical spindle cells and pleomorphic epithelioid cells. Images of mitosis and multinucleated giant cells are common. Cells typically display a fascicular pattern and less frequently a storiform one, although nonspecific patterns may be observed (Fig. 5). Some degree of inflammatory infiltration is often seen, as is evidence of hemorrhaging with hemosiderin deposits. These histologic features are also seen in atypical fibroxanthoma and therefore the 2 tumors are normally indistinguishable.29 The histologic criteria that define PDS and set it apart from atypical fibroxanthoma are subcutaneous tissue invasion, perineural or perivascular invasion, and foci of necrosis. Any of these findings in a cutaneous tumor with histologic features of atypical fibroxanthoma are sufficient to establish a diagnosis of PDS.29,31,33–35
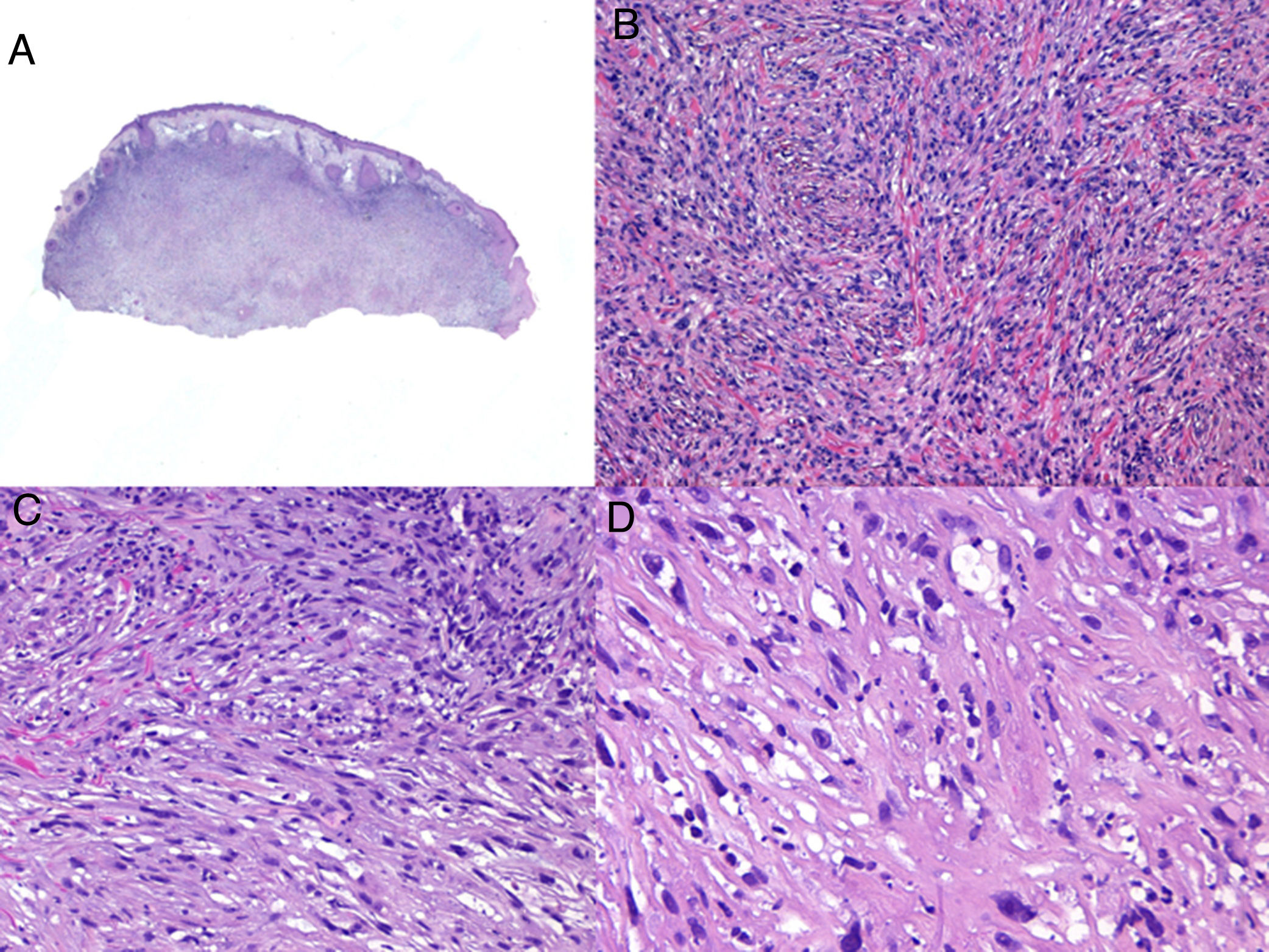
According to the 2 largest series of PDS to date,33,34 15% to 16% of tumors had invaded the subcutaneous tissue, while 61% to 75% had invaded the fascia or underlying muscle, clearly reflecting the aggressive nature of this tumor. Many cases of metastatic atypical fibroxanthoma described in the literature were probably actually PDS.36 Perineural invasion was observed in 28% of the tumors described by Miller et al.34 and in none of those described by Tardío et al.33 Vascular invasion and necrosis, in turn, were observed in 17% to 26% and 17% to 53% of PDSs, respectively.33,34 There are no specific immunohistochemical markers for PDS, although positive results are observed for vimentin, CD10, CD99, and actin. These, however, are all nonspecific markers and serve only to guide diagnosis.37 It is important, however, and more useful to request the inclusion of markers that are negative in PDS and positive in other entities contemplated in the differential diagnosis. A cytokeratin panel should be ordered to rule out poorly differentiated squamous cell carcinoma. This panel should include high-molecular-weight cytokeratins, as certain sarcomatoid or spindle cell squamous cell carcinomas may test negative to cytokeratins with a low molecular weight. The melanocytic markers protein S100, HMB-45, and Melan-A should be ordered to rule out spindle cell or desmoplastic melanoma.29 It should be noted, however, that foci of S100-positive dendritic cells can be observed in PDS. The presence of multinucleated giant cells may also cause focal positivity for Melan-A. When faced with a differential diagnosis featuring PDS and angiosarcoma, note that CD31 positivity may be seen in some cases of PDS.33,34 In this case, the vascular stains, CD31, ERG, and CD34, will be positive in angiosarcoma and negative in PDS. CD34 also helps to distinguish between PDS and dermatofibrosarcoma protuberans.34
ManagementAs PDS is a recently defined tumor, there are no standardized guidelines for its management.
Treatment is surgical and is normally curative. The main risk factor for recurrence is the presence of positive or borderline surgical margins,33,34 hence the general recommendation for wide excision with margins of at least 1cm.38 Because PDS invades the subcutaneous tissue and even the fascia or muscle in up to 75% of cases, meticulous surgery with negative deep margins is crucial. The slow Mohs technique, which allows more rigorous analysis of margins, is recommended for recurrent tumors, tumors in difficult-to-access locations, and tumors suspected to have unpredictable subclinical extension. The benefits of this technique, however, have not yet been studied in this setting.
Preoperative imaging studies are not normally necessary in PDS. If deep invasion is suspected, a CT study should be performed to investigate bone involvement and MRI to investigate deep soft tissue involvement.
Staging is not normally necessary in PDS. Imaging studies should be ordered, following evaluation of the patients’ history, for cases consisting of large tumors, long-standing tumors, or multiple recurrences. In the series described by Tardió et al.,33 recurrence was more common among larger tumors and size should therefore be considered one of the main risk factors for metastasis.
Similarly to with other sarcomas, radiation therapy should be reserved for inoperable cases of PDS or for palliative treatment.
Classic chemotherapy with adriamycin or ifosfamide is used in patients with metastasis.34
PrognosisBased on the 2 series published to date, 20% to 28% of PDS cases recur and 10% to 20% metastasize, mainly to the skin, lungs, or lymph nodes.33,34
Follow-upFollow-up visits consisting of clinical examination of the skin and lymph node stations should be scheduled every 3 months for the first year and every 6 months for the next 4 years. Thereafter, depending on the case, patients could be scheduled for annual check-ups up to year 10.
Recommended follow-up tests are blood tests and chest radiography for patients with an increased risk of distant metastasis due to tumor size, time since onset, or deep invasion.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Llombart B, Serra-Guillén C, Requena C, Alsina M, Morgado-Carrasco D, Machado I, et al. Leiomiosarcoma y sarcoma pleomórfico dérmico: directrices para el diagnóstico y tratamiento. Actas Dermosifiliogr. 2019;110:4–11.


