







El leiomiosarcoma de la piel se clasifica en tres grupos: dérmico, hipodérmico y cutáneo metastásico. El dérmico se origina de las fibras musculares lisas del músculo erector del pelo, dartos genital o de la areola mamaria. Se considera un tumor de malignidad intermedia, con tendencia a la recidiva local (24%) y un bajo riesgo de metástasis (4%). El leiomiosarcoma hipodérmico se origina de las paredes musculares de los vasos, y se caracteriza por presentar una mayor tasa de recidiva local (37%) y metástasis (43%).
El sarcoma pelomórfico dérmico aparece habitualmente en pacientes ancianos y se localiza característicamente en zonas de piel fotoexpuesta (cuero cabelludo). Comparte características histológicas e inmunohistoquímicas con el fibroxantoma atípico, pero con un comportamiento más agresivo (metástasis en el 10-20%). Los criterios histológicos que lo diferencian son la infiltración del tejido celular subcutáneo, la infiltración perineural y la presencia de focos de necrosis.
There are 3 types of leiomyosarcoma of the skin: dermal, subcutaneous, and metastatic cutaneous. Dermal leiomyosarcoma arises from smooth muscle fibers in arrector pili muscles, genital dartos muscles, and the nipple-areola complex. It is an intermediate-grade tumor associated with a tendency for local recurrence (24%) and low metastatic potential (4%). Subcutaneous leiomyosarcoma originates from smooth muscle in blood vessel walls and has higher rates of local recurrence (37%) and metastasis (43%).
Plemorphic dermal sarcoma typically affects elderly patients and arises in sun-exposed areas (e.g., the scalp). Its histologic and immunohistochemical characteristics are similar to those of atypical fibroxanthoma, but it is more aggressive (metastasis rate of 10-20%). Histologically, it can be distinguished from atypical fibroxanthoma by the observation of subcutaneous tissue invasion, perineural invasion, and foci of necrosis.
El leiomiosarcoma (LMS) es una neoplasia de estirpe muscular que nace en los tejidos blandos profundos, útero o más raramente en la dermis. Representa entorno al 5-10% de todos los sarcomas pero solo el 2-3% de los sarcomas cutáneos (0,04% de todas las neoplasias cutáneas)1,2. En la piel es el tercero en frecuencia, por detrás del dermatofibrosarcoma y el sarcoma de Kaposi. Los LMS del retroperitoneo y de la cavidad abdominal son el subgrupo más frecuente y su evolución es mucho más agresiva.
Tradicionalmente, el LMS de la piel se clasifica en tres grandes grupos con diferente implicación pronóstica: LMS cutáneo o dérmico, LMS subcutáneo o hipodérmico y LMS metastásico3. El pronóstico es peor cuanto más profunda es la lesión. El LMS dérmico se origina de las fibras musculares lisas del músculo erector del pelo, el músculo dartos genital o el músculo de la areola mamaria, mientras que el LMS hipodérmico se origina de las paredes musculares de los vasos4. Ambos pueden progresar dando lugar a metástasis y estas pueden aparecer en la piel. No obstante, si encontramos una metástasis en piel de LMS lo más frecuente es que el origen sea de un LMS del retroperitoneo.
El LMS cutáneo dérmico se considera un tumor de malignidad intermedia, con tendencia a la recidiva local (24%) y un bajo riesgo de metástasis (4%)5. Es por ello que Kraft et al.6 propusieron que los LMS dérmicos con mínima afectación hipodérmica no deberían considerarse sarcomas y sugirieron denominar a estas lesiones «neoplasias atípicas intradérmicas del músculo liso». Sin embargo, este término no ha sido ampliamente aceptado. De hecho, el dermatofibrosarcoma protuberans presenta una tendencia mucho menor a producir metástasis cutáneas y no por ello deja de ser un sarcoma. El LMS hipodérmico o subcutáneo se caracteriza por presentar una mayor tasa de recidiva local (37%) y metástasis a distancia (43%). Por último, las metástasis cutáneas de LMS son un indicador de progresión del tumor primario, generalmente de origen visceral, con una esperanza de vida de aproximadamente 16 meses tras la detección de la metástasis7.
Es importante resaltar que antes de realizar el diagnóstico definitivo de un LMS primario de la piel hay que descartar la posibilidad de un LMS metastásico en la piel a partir de un LMS de tejidos profundos o viscerales, sobre todo, si se trata de un LMS subcutáneo.
Características clínicasEl LMS se ha descrito en cualquier rango de edad, pero la mayoría de los casos se desarrollan en pacientes de edad avanzada adulta, con un pico de incidencia entre los 50-70 años. Aparece más frecuentemente en hombres con una relación 3:1 y parece presentarse más en la raza caucásica3. La mayoría de los LMS de la piel se originan de novo y no de una lesión previa tipo piloleiomioma. Recientemente se ha identificado una alteración del gen de la fumarato hidratasa en algunos LMS de pacientes con leiomiomatosis hereditaria y cáncer renal familiar8. Han sido descritos LMS en zonas tratadas previamente con radioterapia y algunos pacientes refieren un antecedente de traumatismo o cicatriz, sin embargo, en la mayoría de los pacientes no hay un factor previo desencadenante conocido.
El 50% de los LMS cutáneos se localizan preferentemente en la superficie extensora de las extremidades inferiores, y menos frecuentemente en cuero cabelludo y cara9, aunque también se han descrito casos en tronco, labio, región genital (escroto, vulva, pene) y glútea.
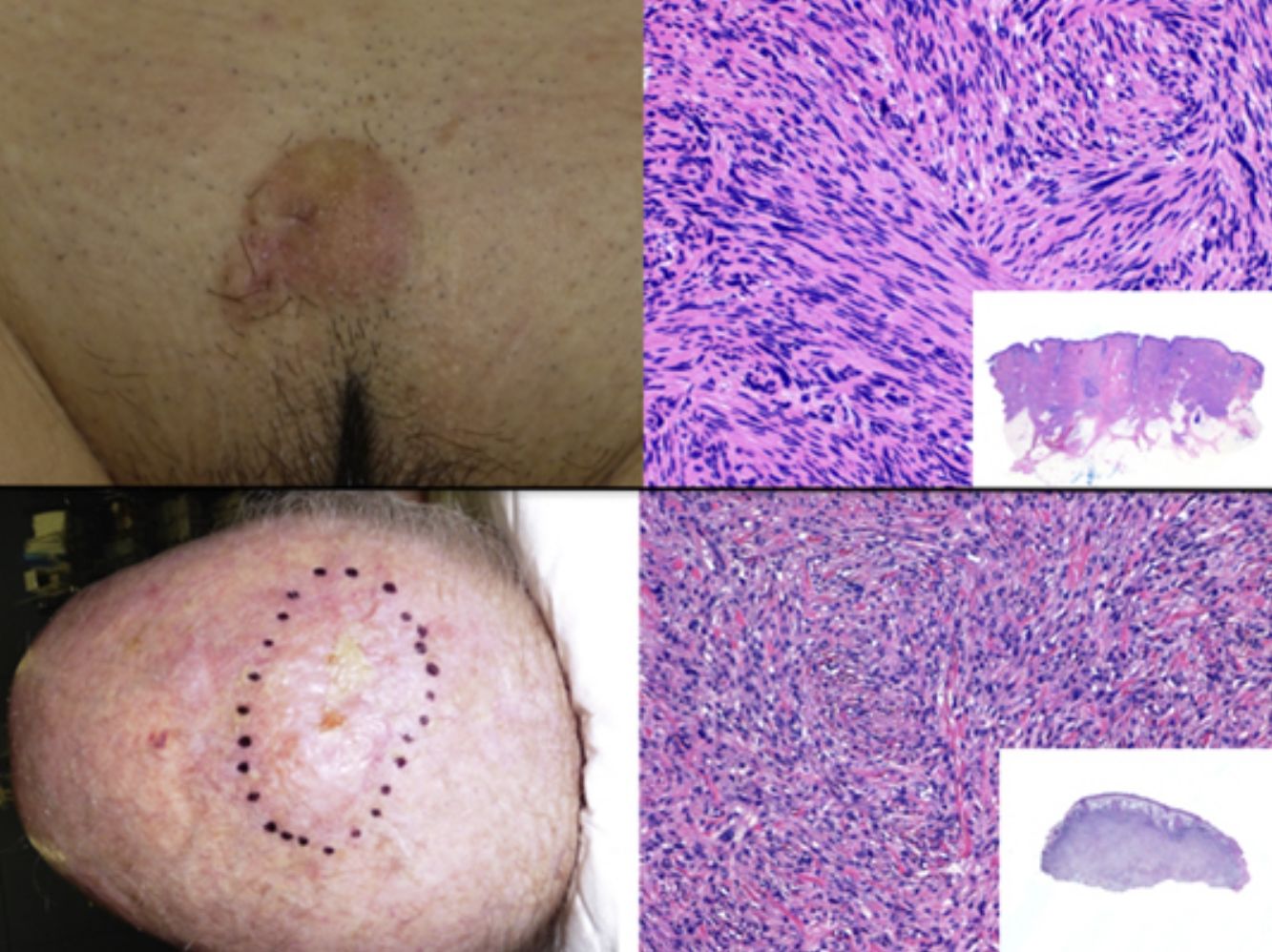
La presentación clínica del LMS es inespecífica. Lo más frecuente es un único nódulo firme, de superficie lisa y rosada o bien una tumoración más exofítica de coloración rojiza o marrón (fig. 1). Las lesiones subcutáneas clínicamente parecen mejor delimitadas que las dérmicas y recuerdan a un lipoma pero con una consistencia más sólida. Se han descrito LMS tipo placa con múltiples nódulos agrupados y muy indurados. La lesión dérmica suele ser de crecimiento lento con un tamaño al momento del diagnóstico entre 1 y 3,5cm (0,5 a 19cm), y generalmente son más grandes los LMS subcutáneos. Es muy frecuente que la lesión duela a la palpación (63%) y más raro de manera espontánea (25%)9. Otros síntomas asociados son prurito, ardor y parestesias3,9.
Características histopatológicas
Es imprescindible para el diagnóstico realizar una biopsia que incluya tejido celular subcutáneo.
En el estudio histológico, el LMS dérmico suele ser una lesión mal delimitada que ocupa todo el espesor de la dermis e infiltra en ocasiones al tejido celular subcutáneo (fig. 2a y b.). El LMS subcutáneo es una lesión mejor delimitada, circunscrita, comprime el tejido adyacente y se localiza enteramente en la hipodermis respetando la dermis. En ambos casos, a pequeño aumento, la lesión parece constituida por fascículos entrelazados de fibras musculares lisas. Las células son fusiformes, con núcleos de forma elongada y extremos romos, nucleolo poco llamativo y citoplasma eosinófilo fibrilar (fig. 2 c y d). Algunas células presentan un halo claro perinuclear característico de la célula muscular4,10,11.
 Visión panorámica de una tumoración dérmica mal delimitada que infiltra al tejido celular subcutáneo. b) A mayor detalle la formación en dermis de fascículos de células fusiformes entrelazadas y distribuidos irregularmente por la dermis y que recuerdan a fibras musculares. c) La tumoración infiltra el tejido celular subcutáneo. d) Fascículos de células fusiformes pleomórficas que se entrecruzan en ángulo recto y la presencia de figuras mitósicas.")
Histología típica de un leiomiosarcoma. a) Visión panorámica de una tumoración dérmica mal delimitada que infiltra al tejido celular subcutáneo. b) A mayor detalle la formación en dermis de fascículos de células fusiformes entrelazadas y distribuidos irregularmente por la dermis y que recuerdan a fibras musculares. c) La tumoración infiltra el tejido celular subcutáneo. d) Fascículos de células fusiformes pleomórficas que se entrecruzan en ángulo recto y la presencia de figuras mitósicas.
Desde el punto de vista histopatológico, se han descrito dos patrones de crecimiento, nodular y difuso12. El patrón nodular se caracteriza por una mayor celularidad, atipia y un mayor número de mitosis, y el patrón difuso, por una menor celularidad, menor pleomórfismo y un menor número de mitosis13.
Las lesiones dérmicas con patrón de crecimiento difuso presentan poca atipia celular por lo que puede ser difícil diferenciarlas del leiomioma. Si bien un patrón más infiltrativo y una mayor celularidad favorecen el diagnóstico de malignidad, el hallazgo de alguna figura mitósica es clave para su confirmación.
Se ha descrito una serie de variantes histopatológicas de LMS como el LMS de células epitelioides14, el LMS de células gigantes multinucleadas15, el LMS de células granulares16, y el LMS esclerótico17; asimismo existen casos que presentan gran desmoplasia, lo que puede dificultar el diagnóstico. En lesiones subcutáneas se han descrito las variantes mixoides y pleomórficas4.
Ante la sospecha histológica de un LMS es necesario realizar un estudio inmunohistoquímico que nos ayude a descartar otros tumores fusocelulares con características histológicas similares. Los LMS bien diferenciados presentan positividad a la vimentina, la desmina, el h-caldesmon, la actina muscular específica, la actina alfa de músculo liso y miosina del músculo liso4. En lesiones más indiferenciadas y en LMS subcutáneos la desmina es frecuentemente negativa18,19. En el LMS son necesarios al menos dos marcadores inmunohistoquímicos de estirpe muscular para poder confirmar el diagnóstico. En algunas ocasiones la proteína S-100 es positiva, al igual que las citoqueratinas. Otras tinciones inmunohistoquímicas realizadas para descartar otras lesiones fusocelulares (carcinoma fusocelular, melanoma desmoplásico, dermatofibrosarcoma protuberans, tumor maligno de la vaina nerviosa periférica y tumores vasculares) son: EMA, CD34, CD117, CEA, HMB45, Mart-1, Melan A y CK7, y todas ellas son negativas en el LMS.
EstadificaciónActualmente no existe un TNM específico para los LMS, de modo que se emplea el TNM de los sarcomas de partes blandas de la clasificación de la AJCC.
El pronóstico del LMS depende de si se trata de una lesión cutánea o subcutánea. El LMS dérmico tiene tendencia a la recidiva local (24%) pero poca probabilidad de metástasis (4%). El LMS subcutáneo tiene más tendencia a la recidiva local (37%) y a las metástasis (43%). Por este motivo, es lógico que la estatificación y el seguimiento sean diferentes.
LMS dérmico
Es recomendable realizar una resonancia magnética de la zona previa a la cirugía, sobre todo en las lesiones de gran tamaño, infiltradas a la palpación o en localizaciones difíciles de acceder (cabeza). Aunque no existen estudios, es muy posible que la ecografía sea una ayuda al dermatólogo y pueda sustituir a la resonancia magnética (fig. 3). Además, se debería incluir una radiografía de tórax previa a la cirugía.

LMS subcutáneo
Es recomendable siempre realizar una resonancia magnética de la zona previa a la cirugía y una TC toracoabdominal para descartar la posibilidad de un LMS metastásico en la piel a partir de un LMS de tejidos profundos20(fig. 3).
Tratamiento y pronósticoDada la escasa incidencia del LMS en piel es recomendable que el tratamiento se realice en un centro especializado en sarcomas.
La extirpación quirúrgica completa es el tratamiento de elección para el LMS. Los mejores resultados se obtienen mediante cirugía local amplia o mediante control de márgenes con cirugía micrográfica de Mohs (CMM).
El principal dilema en la cirugía de los LMS son la amplitud de los márgenes quirúrgicos pues no están bien establecidos en la literatura. Antiguamente las recomendaciones eran cirugías muy amplias con márgenes de 3-5cm, sin embargo, hoy en día se han obtenido resultados similares con cirugías más conservadoras con márgenes de 1-3cm3,21–24 (fig. 3). El margen profundo debe alcanzar hasta la fascia, y en casos más infiltrantes deberá incluirse el músculo.
La experiencia del tratamiento de LMS cutáneo con la CMM es escasa. Se han publicado unos 50 casos tratados con CMM, siendo el porcentaje de recidivas de entre 0 y 13%3,22,23,25. Este porcentaje es mucho menor que el observado en los LMS tratados con cirugía convencional, que varía entre 9 y 40% de recidivas3,21–23, por lo que la CMM parece ser una buena opción quirúrgica en los LMS superficiales dérmicos.
El papel de la radioterapia en el LMS cutáneo todavía es discutido. La radioterapia adyuvante puede tener especial relevancia cuando los márgenes quirúrgicos están afectados o son muy ajustados tras la extirpación y no es posible una nueva resección, en particular en las lesiones profundas y las de grado alto. Los factores asociados a un peor pronóstico son: tumoración subcutánea, tamaño de la tumoración mayor de 5cm, localización acral, aneuploidia celular y el hallazgo de invasión vascular13. La radioterapia también se puede utilizar en el control local paliativo en casos con metástasis.
En el tratamiento de enfermedad metastásica, la quimioterapia es fundamental. Los agentes más utilizados incluyen la doxorrubicina e ifosfamida, gemcitabina y taxotere, dacarbacina y trabectedina. Estos tratamientos no son curativos, pero pueden retrasar la progresión de la enfermedad26.
SeguimientoNo existen guías estandarizadas en el seguimiento de LMS, pero se recomienda realizar un examen clínico cada 4 meses durante los dos primeros años para la detección precoz de posibles recidivas locales. Se aconseja a partir de entonces controles cada 6 meses hasta el quinto año después de la cirugía y posteriormente, una vez al año hasta los 20 años, ya que se han descrito recidivas muy tardías3,11. Tampoco existe una guía estandarizada de las exploraciones radiológicas a realizar en el seguimiento postoperatorio de los pacientes con sarcomas cutáneos (fig. 3). Sin embargo, la práctica de una radiografía simple de tórax anual los cinco primeros años tras la cirugía y la evaluación clínica del lecho quirúrgico y de los ganglios linfáticos locorregionales parece una estrategia correcta26. En algunas ocasiones, la RM puede ser de ayuda, sobre todo en lesiones recurrentes o hipodérmicas o aquellos casos que la cirugía haya sido compleja.
La tasa de metástasis en los LMS dérmicos es del 4% (3-14%) y en los LMS subcutáneos del 43% (21-62%)3. Las metástasis se producen fundamentalmente por vía hematógena al pulmón, la piel y menos frecuentemente, a los ganglios linfáticos regionales. La prueba de estatificación idónea para pacientes con sospecha o control de enfermedad diseminada es la TC helicoidal toracoabdominal26.
Sarcoma pleomórfico dérmicoIntroducciónEl sarcoma pleomórfico indiferenciado, antiguamente denominado histiocitoma fibroso maligno es un tumor maligno de partes blandas (sarcoma), característicamente muy pleomórfico, en el que no se encuentra una línea de diferenciación mediante técnicas inmunohistoquímicas27–32.
Al sarcoma pleomórfico indiferenciado que se inicia en la piel se le denomina sarcoma pleomórfico dérmico (SPD), un tumor con unas características histológicas comunes al fibroxantoma atípico (FXA) pero con un peor pronóstico33,34. Debido a la confusión que históricamente se ha creado en la nomenclatura de estos tumores y a la actualización llevada a cabo por la Organización Mundial de la Salud31 en 2013, existen pocas series amplias de SPD de las que se puedan extraer experiencia y conclusiones para el adecuado manejo de este tumor.
DefiniciónEl SPD es un tumor cutáneo de histogénesis incierta, que muestra unas características clínicas poco específicas y comparte características histológicas e inmunohistoquímicas con el FXA, pero con un comportamiento más agresivo que este29.
Presentación clínicaEl SPD aparece habitualmente en pacientes ancianos y se localiza característicamente en zonas de piel fotoexpuesta, predominantemente en la cabeza (fig. 4) y sobre todo en el cuero cabelludo. En este sentido, tal y como señalan Tardío et al., se debe descartar otros tumores y dudar del diagnóstico de SPD si no aparece en piel dañada por el sol de pacientes mayores33. El SPD se manifiesta como un tumor exofítico, asimétrico, comúnmente ulcerado y sangrante, de crecimiento rápido y de un tamaño medio de 2,2 a 2,5cm33,34, aunque hay casos de pocosmm y otros de varios cm. Clínicamente sería indistinguible del FXA, pero con una extensión subclínica mayor, de modo que habitualmente no se sospecha clínicamente y suele diagnosticarse de carcinoma epidermoide.
Características histopatológicas y diagnóstico
Al tratarse de un tumor cuyo origen permanece desconocido y no se conoce la célula de la que deriva, el diagnóstico es de exclusión.
El SPD está localizado en la dermis, sin conexión con la epidemis y sin presencia de zona Grenz (zona de colágeno normal en la dermis que separa la epidermis del tumor)33,34. Está formado por dos poblaciones de células en diferente proporción, unas fusiformes atípicas y otras epitelioides pleomórficas. Es frecuente encontrar imágenes de mitosis y células gigantes multinucleadas. Las células se disponen habitualmente formando un patrón fascicular, con menos frecuencia estoriforme, aunque pueden disponerse sin un patrón específico (fig. 5). También es frecuente observar cierto grado de infiltrado inflamatorio y de hemorragia con depósitos de hemosiderina. Estos hallazgos histológicos descritos para el SPD coinciden con los del FXA por lo que habitualmente son tumores indistinguibles29. Los criterios histológicos que definen al SPD y lo diferencian de FXA son: la infiltración del tejido celular subcutáneo (TCS), la infiltración perineural o perivascular y la presencia de focos de necrosis. Cualquiera de estos hallazgos en el seno de un tumor cutáneo con características histológicas de FXA son suficientes para diagnosticar un SPD y descartar el FXA29,31,33–35.
 Panorámica de una lesión tumoral que infiltra la dermis e hipodermis. b) Tumor constituido por células fusiformes siguiendo un patrón estoriforme. c) Las células se encuentran inmersas en un estroma mixoide. d) Detalle de algunas células más pleomórficas con citoplasma amplio.")
Histología de un sarcoma pleomórfico. a) Panorámica de una lesión tumoral que infiltra la dermis e hipodermis. b) Tumor constituido por células fusiformes siguiendo un patrón estoriforme. c) Las células se encuentran inmersas en un estroma mixoide. d) Detalle de algunas células más pleomórficas con citoplasma amplio.
En las dos series de referencia de SPD, los tumores afectaron al TCS en un 15-16% y a la fascia o músculo subyacente en un 61-75% lo que traduce ciertamente la agresividad del SPD33,34. Probablemente muchos de los casos de FXA metastásicos descritos en la literatura corresponden realmente a SPD36. La infiltración perineural se encontró en el 28% de los tumores en la serie de Miller34 y en ningún caso en la serie de Tardío33. La infiltración vascular se encontró en un 17-26% de los casos y la necrosis en un 17-53% de los SPD33,34. Desde el punto de vista de la inmunohistoquímica el SPD no tiene un marcador característico, aunque es positivo para la vimentina, el CD10, el CD99 y la actina, todos ellos son inespecíficos y tienen un valor orientativo37. Sin embargo, es muy importante y más útil para el diagnóstico, pedir otros marcadores que resultan negativos en el SPD y positivos en otros tumores con los que hay que hacer el diagnóstico diferencial. Hay que solicitar un panel de citoqueratinas (CK) para descartar un carcinoma espinocelular pobremente diferenciado que incluya las CK de alto peso molecular, puesto que algún caso de carcinoma espinocelular sarcomatoide o de células fusiformes, las CK de bajo peso molecular, podrían ser negativas. También es necesario solicitar marcadores melanocíticos como la proteína S100, el HMB-45 y melan-A para descartar melanoma de células fusiformes o el desmoplásico29, sin embargo, hay que tener en cuenta que se pueden encontrar focos positivos de S100 en el seno de un SPD debido a la presencia de células dendríticas y además también es posible encontrar expresión focal de melan-A correspondiente a las células gigantes multinucleadas. Para el diagnóstico diferencial entre el SPD y el angiosarcoma hay que tener en cuenta que el CD31 puede ser positivo en algunos casos de SPD33,34. Por lo tanto, habría que completar el panel de marcadores vasculares con el CD31, ERG, CD34, que resulta negativo en el SPD y positivo en el angiosarcoma. Además, este último marcador también resultaría necesario en el hipotético caso de establecerse un diagnóstico diferencial entre el SPD y el dermatofibrosarcoma protuberans34.
ManejoTeniendo en cuenta que el SPD es un tumor de reciente definición no existen guías de consenso para su manejo.
El tratamiento del SPD es quirúrgico y habitualmente es curativo. El factor que más se correlaciona con la posibilidad de recidiva es la presencia de márgenes quirúrgicos positivos o ajustados33,34 por lo tanto la recomendación general sería realizar una cirugía amplia con al menos 1cm de margen38. Teniendo en cuenta que por definición el SPD invade el TCS y que puede llegar a infiltrar la fascia o el músculo hasta en el 75% de los casos, es muy importante realizar una cirugía lo suficientemente meticulosa para asegurar un margen profundo negativo. En los casos recidivados o en aquellos de localización comprometida o con sospecha de extensión subclínica impredecible sería recomendable realizar CMM diferida para un análisis más riguroso de los márgenes. sin embargo, actualmente no existen trabajos que lo avalen.
Habitualmente no es preciso solicitar pruebas de imagen preoperatorias. En los casos en los que se sospeche infiltración profunda se solicitará una TC para descartar afectación ósea o una resonancia magnética para estudiar la afectación profunda de partes blandas.
En la mayoría de los casos de SPD no está indicado solicitar estudio de extensión. Las pruebas de imagen se deberán solicitar, basándose en la anamnesis, en aquellos casos que por su tamaño, tiempo de evolución o presentación clínica en forma de múltiples recidivas lo precisen. En este sentido, tal y como se observa en la serie de Tardío y cols.33 los tumores que recidivaron en su serie fueron precisamente aquellos de mayor tamaño, por lo tanto, el tamaño debe ser uno de los factores más relacionados en las metástasis.
El papel de la radioterapia en el tratamiento del SPD, al igual que en otros sarcomas, se reserva para casos inoperables o para aquellos en los que se decida un tratamiento paliativo.
En los casos metastásicos se emplearía la quimioterapia clásica con adriamicina o ifosfamida34.
PronósticoSegún las dos series publicadas hasta la fecha el SPD recidiva en un 20-28% de los casos y metastatiza en el 10-20% de los casos, sobre todo a piel, pulmón y ganglios linfáticos33,34.
SeguimientoSe recomienda un seguimiento mediante exploración clínica de la piel y de los territorios ganglionares cada 3 meses el primer año y posteriormente cada 6 meses hasta completar 5 años de seguimiento. Posteriormente, según el caso, se podría revisar al paciente una vez al año hasta 10 años tras la intervención.
Estaría indicada una analítica y una radiografía de tórax en aquellos casos que por su tamaño, tiempo de evolución o infiltración en profundidad tuvieran un mayor riesgo de hacer metástasis a distancia en los seguimientos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



