





La vía Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) es esencial en la señalización final de una gran mayoría de interleucinas (IL) fundamentales en la patogénesis de la dermatitis atópica (DA). El bloqueo transversal que consiguen los inhibidores de JAK a través de la inhibición intermitente de las acciones de múltiples citoquinas, permite modular la inflamación Th2, la disfunción de barrera epidérmica y la señalización del prurito. Sin embargo, esa inhibición amplia también puede asociarse con una mayor variedad de efectos adversos. En este artículo se revisan los inhibidores de JAK recientemente aprobados en la DA —baricitinib, upadacitinib y abrocitinib—, así como otros emergentes o en desarrollo como gusacitinib, delgocitinib, ruxolitinib, brepocitinib, tofacitinib y cerdulatinib. El bloqueo de la señalización de diversas citoquinas relevantes en esta dermatosis, compleja patogénicamente y con una expresión fenotípica heterogénea, a través de los inhibidores de JAK, ha supuesto una revolución en el tratamiento de la DA.
The JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathway is an essential final step in the signaling process of most interleukins with a critical role in the pathogenesis of atopic dermatitis. By achieving broad, intermittent inhibition of the activity of multiple cytokines, JAK inhibitors help modulate T helper 2 cell–mediated inflammation, epidermal barrier dysfunction, and itch signaling. This comprehensive blockade, however, can result in a wider range of adverse effects. We review a number of JAK inhibitors that have been recently approved for use in atopic dermatitis, such as baricitinib, upadacitinib, and abrocitinib, as well as others that are currently in the pipeline or under development, such as gusacitinib, delgocitinib, ruxolitinib, brepocitinib, tofacitinib, and cerdulatinib. The use of JAK inhibitors to block the signaling of numerous cytokines with a critical role in the pathogenesis of atopic dermatitis has revolutionized the treatment of this pathogenically complex, phenotypically heterogeneous skin disease.
La dermatitis atópica (DA) es una enfermedad inflamatoria crónica y recurrente1, asociada a una gran morbilidad2. Se trata de una enfermedad heterogénea en sus fenotipos clínicos, lo que refleja una diversidad de mecanismos fisiopatológicos subyacentes, así como la interacción entre la predisposición genética, los factores ambientales y la desregulación inmunológica3. En la última década, se han producido grandes avances en el conocimiento de su patogénesis, circunstancia que ha permitido el desarrollo de nuevas terapias4. La inflamación de la DA está mediada predominantemente por una respuesta Th2, que incluye la participación de las interleucinas (IL)-4, IL-13, IL-33, y también la IL-31, siendo esta última un potente mediador en el prurito. En etapas crónicas de la enfermedad, también existe una participación relevante de la IL-17, IL-22 e interferón-gamma (IFN-γ). Para llevar a cabo sus efectos finales sobre las células, la mayoría de citoquinas implicadas en la DA tienen que actuar sobre un receptor transmembrana: los receptores de citoquinas tipo I y II. Tras la unión de la citoquina a su receptor, se produce la autofosforilación de 2 isoformas de JAK (como homo- o heterodímero), lo que activa a las Signal Transducer and Activator of Transcription (STAT), que se translocan finalmente al núcleo donde modulan la transcripción de los genes diana3,5.
La aprobación del primer biológico en la DA, dupilumab, dirigido frente a la subunidad alfa del receptor de la IL-4, ha supuesto un salto cualitativo en el tratamiento de la DA6. Recientemente, ha sido aprobado el uso de un anti-IL-13, tralokinumab, y se encuentran en etapas muy avanzadas de su desarrollo clínico otros anticuerpos monoclonales7 como lebrikizumab (un inhibidor de la señalización de IL-13), con buenas perspectivas de respuesta clínica en signos y síntomas8,9 y nemolizumab (anti-IL31), que consigue una rápida y sostenida reducción del prurito, sin alcanzar, sin embargo, mejorías tan significativas del eccema en comparación a las terapias innovadoras actualmente disponibles10,11. Por otro lado, teniendo en cuenta la heterogeneidad clínica entre pacientes y, en ocasiones, la variable expresión fenotípica en el curso de la enfermedad en el mismo individuo, a la que probablemente subyace la gran diversidad de citoquinas implicadas en los procesos patogénicos de la DA, existe un interés creciente en los inhibidores de las Janus Kinasa (iJAK), moléculas que interfieren, simultáneamente, en la señalización de múltiples citoquinas.
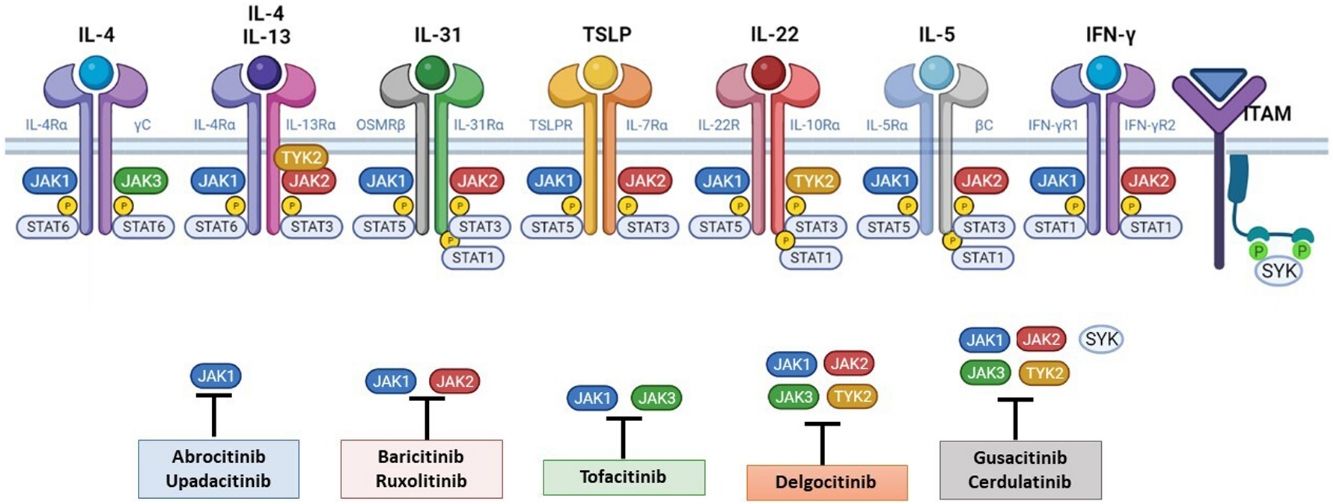
El papel de la vía JAK-STAT en la patogénesis de la dermatitis atópicaEstructura y función de la vía de señalización JAK-STATEn los últimos años se han desarrollado moléculas eficaces en la DA, que actúan modulando la actividad de la vía de señalización JAK/STAT, esencial en la inmunidad innata y adaptativa12. Para que las citoquinas implicadas en la DA tengan sus efectos finales sobre las células, deben actuar a través de su receptor transmembrana, los receptores de citoquinas de tipo I y II. Estos receptores no disponen de actividad tirosin-kinasa intrínseca, y precisan de la acción de las enzimas citoplasmáticas JAK (tabla 1)13,14. Se han identificado 4 subunidades de JAK en las células humanas: JAK1, JAK2, JAK3 y TYK2. Estas forman entre sí heterodímeros y heterotrímeros; JAK2 además puede formar homodímeros (fig. 1)12,15. Cada receptor de citoquinas recluta y utiliza una combinación particular de JAK, circunstancia con implicación en el uso terapéutico de la inhibición de JAK en distintas enfermedades inmunomediadas y neoplasias16.
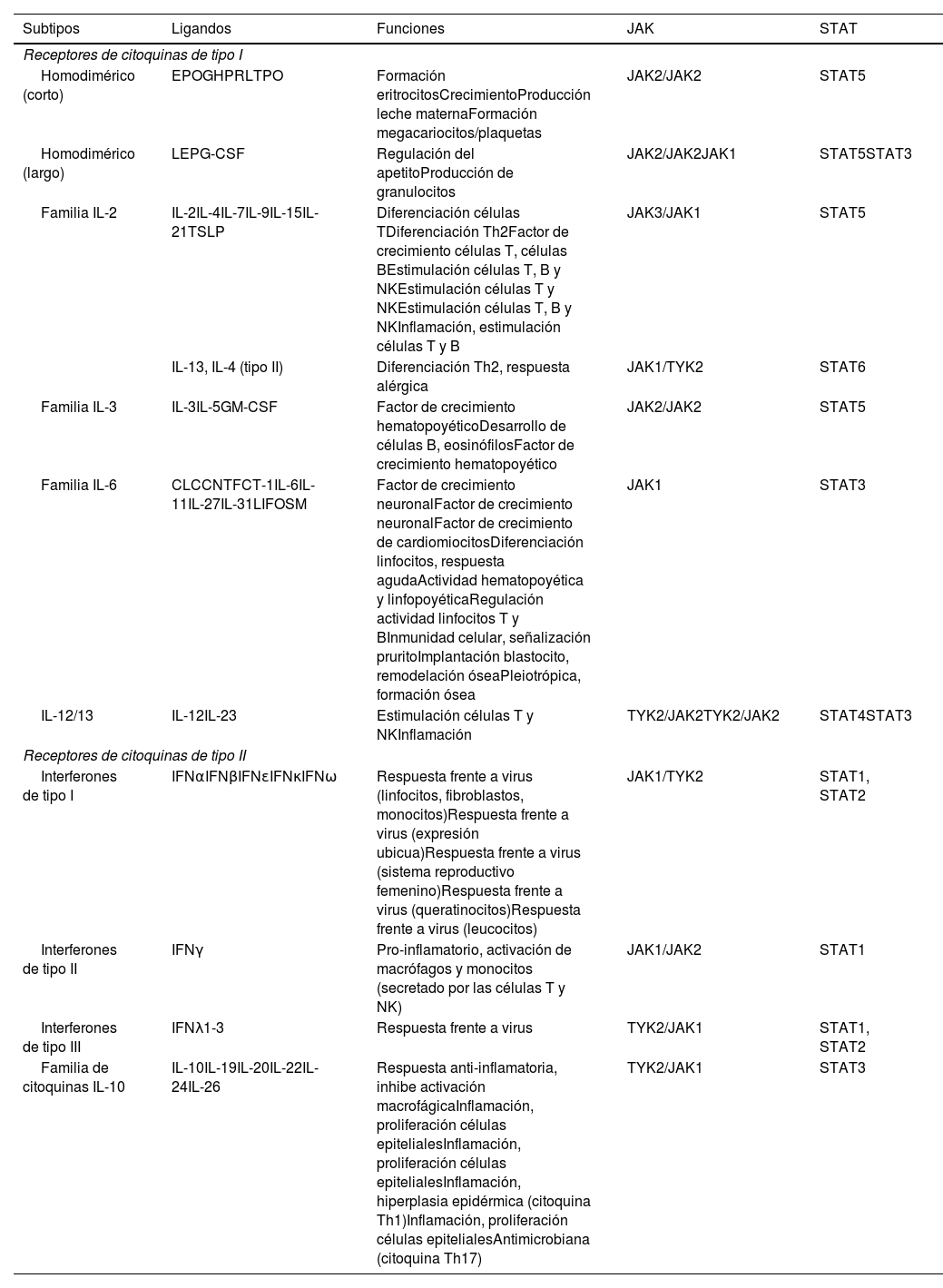
Receptores de citoquinas de tipo I y II
| Subtipos | Ligandos | Funciones | JAK | STAT |
|---|---|---|---|---|
| Receptores de citoquinas de tipo I | ||||
| Homodimérico (corto) | EPOGHPRLTPO | Formación eritrocitosCrecimientoProducción leche maternaFormación megacariocitos/plaquetas | JAK2/JAK2 | STAT5 |
| Homodimérico (largo) | LEPG-CSF | Regulación del apetitoProducción de granulocitos | JAK2/JAK2JAK1 | STAT5STAT3 |
| Familia IL-2 | IL-2IL-4IL-7IL-9IL-15IL-21TSLP | Diferenciación células TDiferenciación Th2Factor de crecimiento células T, células BEstimulación células T, B y NKEstimulación células T y NKEstimulación células T, B y NKInflamación, estimulación células T y B | JAK3/JAK1 | STAT5 |
| IL-13, IL-4 (tipo II) | Diferenciación Th2, respuesta alérgica | JAK1/TYK2 | STAT6 | |
| Familia IL-3 | IL-3IL-5GM-CSF | Factor de crecimiento hematopoyéticoDesarrollo de células B, eosinófilosFactor de crecimiento hematopoyético | JAK2/JAK2 | STAT5 |
| Familia IL-6 | CLCCNTFCT-1IL-6IL-11IL-27IL-31LIFOSM | Factor de crecimiento neuronalFactor de crecimiento neuronalFactor de crecimiento de cardiomiocitosDiferenciación linfocitos, respuesta agudaActividad hematopoyética y linfopoyéticaRegulación actividad linfocitos T y BInmunidad celular, señalización pruritoImplantación blastocito, remodelación óseaPleiotrópica, formación ósea | JAK1 | STAT3 |
| IL-12/13 | IL-12IL-23 | Estimulación células T y NKInflamación | TYK2/JAK2TYK2/JAK2 | STAT4STAT3 |
| Receptores de citoquinas de tipo II | ||||
| Interferones de tipo I | IFNαIFNβIFNɛIFNκIFNω | Respuesta frente a virus (linfocitos, fibroblastos, monocitos)Respuesta frente a virus (expresión ubicua)Respuesta frente a virus (sistema reproductivo femenino)Respuesta frente a virus (queratinocitos)Respuesta frente a virus (leucocitos) | JAK1/TYK2 | STAT1, STAT2 |
| Interferones de tipo II | IFNγ | Pro-inflamatorio, activación de macrófagos y monocitos (secretado por las células T y NK) | JAK1/JAK2 | STAT1 |
| Interferones de tipo III | IFNλ1-3 | Respuesta frente a virus | TYK2/JAK1 | STAT1, STAT2 |
| Familia de citoquinas IL-10 | IL-10IL-19IL-20IL-22IL-24IL-26 | Respuesta anti-inflamatoria, inhibe activación macrofágicaInflamación, proliferación células epitelialesInflamación, proliferación células epitelialesInflamación, hiperplasia epidérmica (citoquina Th1)Inflamación, proliferación células epitelialesAntimicrobiana (citoquina Th17) | TYK2/JAK1 | STAT3 |
Receptores de citoquinas de tipo I y II. En la tabla se muestran las familias de citoquinas y los receptores a los que se unen, así como las JAK/STAT a través de las cuales se señalizan sus funciones.
CLC: Cardiotrophin cytokine; CNTF: Ciliary NeuroTrophic growth Factor; CT1: Cardiotrophin 1; EPO: eritropoyetina; G-CSF: Granulocyte Colony Stimulating Factor; GH: growth hormone; GM-CSF: Granulocyte/Macrophage Colony Stimulating Factor; IFNα: interferón alfa; IFNβ: interferón beta; IFNɛ: interferón épsilon; IFNκ: interferón kappa; IFNω: interferón omega; IFNγ: interferón gamma; IFNλ1-3: interferones lambda 1, 2 y 3. JAK: Janus Kinasa; LEP: leptina; LIF: Leukemia Inhibitory Factor; OSM: Oncostatin M; PRL: prolactina; STAT: Signal Transducer and Activator of Transcription; TPO: trombopoyetina; TSLP: Thymic Stromal LymphoPoietin; TYK2: Tirosin-kinasa 2.
. Se han identificado más de 50 citoquinas y factores de crecimiento en esta vía de señalización, incluyendo interferones, citoquinas, hormonas y factores estimuladores de colonias. Los eventos posteriores mediados por JAK/STAT incluyen la hematopoyesis, la reparación tisular, la función inmune, la inflamación, la apoptosis y la adipogénesis. Las JAKs están asociadas mediante enlaces no covalentes a los receptores de citoquinas y median la fosforilación de dichos receptores y el reclutamiento de una o más proteínas STAT. Las STAT en su forma fosforilada posteriormente se dimerizan, tanto en forma de homo como heterodímeros, y se translocan al núcleo, donde se unen a secuencias específicas del promotor y regulan la expresión de genes diana, de acuerdo a la acción de las distintas citoquinas. JAK: Janus Kinase; STAT: Signal Transducer and Activator of Transcription; TYK: Tirosin-kinasa 2.Fuente: Figura adaptada de «Cytokine Signaling through the JAK-STAT Pathway», por BioRender.com (2023). Obtenido de https://app.biorender.com/biorender-templates/figures/all/t-5fac3e99614e0c00aac4a356-cytokine-signaling-through-the-jak-stat-pathway")
Se han identificado 4 subunidades de JAK en las células humanas: JAK1, JAK2, JAK3 y TYK2. Estas forman entre sí heterodímeros y heterotrímeros; JAK2, además puede formar homodímeros. Existen 7 tipos de STAT (STAT1-6, incluyendo los homólogos STAT5a y STAT5b). Se han identificado más de 50 citoquinas y factores de crecimiento en esta vía de señalización, incluyendo interferones, citoquinas, hormonas y factores estimuladores de colonias. Los eventos posteriores mediados por JAK/STAT incluyen la hematopoyesis, la reparación tisular, la función inmune, la inflamación, la apoptosis y la adipogénesis. Las JAKs están asociadas mediante enlaces no covalentes a los receptores de citoquinas y median la fosforilación de dichos receptores y el reclutamiento de una o más proteínas STAT. Las STAT en su forma fosforilada posteriormente se dimerizan, tanto en forma de homo como heterodímeros, y se translocan al núcleo, donde se unen a secuencias específicas del promotor y regulan la expresión de genes diana, de acuerdo a la acción de las distintas citoquinas. JAK: Janus Kinase; STAT: Signal Transducer and Activator of Transcription; TYK: Tirosin-kinasa 2.Fuente: Figura adaptada de «Cytokine Signaling through the JAK-STAT Pathway», por BioRender.com (2023). Obtenido de https://app.biorender.com/biorender-templates/figures/all/t-5fac3e99614e0c00aac4a356-cytokine-signaling-through-the-jak-stat-pathway
La patogénesis de la DA es compleja, e implica la intersección de varios aspectos. La respuesta inmune mediada por los linfocitos Th2 es central en la DA, y la vía JAK/STAT participa en la señalización de las citoquinas Th2 (IL-4, IL-13, IL-31). Existen 2 tipos de receptor para la IL-4: el tipo I (formado por la cadena IL-4Rα y la cadena γ común), cuya señalización se lleva a cabo a través de JAK1/JAK3 con la posterior activación de STAT6, y el tipo II (formado por la cadena IL-4Rα y la cadena IL-13Rα1), que lleva a la activación de JAK1 y TYTK2 y la posterior activación de STAT6 y STAT3. El factor de transcripción STAT6 es esencial para las funciones finales de IL-4 e IL-13. Por otro lado, la acción de la IL-31 sobre su receptor IL-31R (formado por la cadena IL-31Rα y la cadena β del receptor de oncostatina M [OSMRβ]) se lleva a cabo a través de la vía JAK/STAT, de la vía de la Protein Kinase B (PKB) y la vía de las Mitogen-Activated Protein Kinase (MAPK). Así, la señalización del prurito, síntoma principal de la enfermedad, se transmite también a través de la vía JAK/STAT17.
A pesar del papel central de la respuesta Th2 en la DA, las respuestas Th1, Th17 y Th22 participan de forma variable en las etapas más crónicas de la enfermedad y de forma más característica en algunos fenotipos4. En particular, la IL-22, perteneciente a la familia IL-10, será fundamental en la hiperplasia epidérmica. Su unión al IL-22R (compuesto por las cadenas IL-22R1 e IL-10R2), induce la activación de JAK1 y TYK2, con activación final de STAT3 (fig. 2). La señalización de IL-17, en cambio, no depende de la vía JAK/STAT.

Principales receptores de citoquinas y su señalización a través de JAK/STAT. Para que las citoquinas implicadas en la DA tengan efectos finales sobre las células, deben unirse a su receptor transmembrana, que precisan de la acción de las enzimas citoplasmáticas JAK. De este modo, la señalización JAK/STAT resulta clave en la patogénesis de la DA. El bloqueo mediante inhibidores de JAK permite una inhibición transversal de las acciones de múltiples citoquinas a la vez. Algunos de ellos bloquen de forma más selectiva alguna de las enzimas JAK, mientras que otros pueden actuar frente a todas sus formas. DA: dermatitis atópica; IFN-γ: interferón-gamma; IL: interleucina; JAK: Janus Kinase; STAT: Signal Transducer and Activator of Transcription; SYK: Spleen Tyrosine Kinase; TSLP: Thymic Stromal Lymphopoietin; TYK: Tirosin-kinasa 2.Fuente: Figura creada con BioRender.com
La estrategia basada en el uso de terapia biológica podría permitir una aproximación dirigida, impidiendo la unión de citoquinas específicas a sus receptores. Teniendo en cuenta que JAK/STAT es la vía de señalización de los receptores de dichas citoquinas, es esperable que los iJAK puedan ofrecer también beneficios terapéuticos en la DA.
La vía JAK-STAT es un paradigma de la rápida señalización de la membrana al núcleo, y los inhibidores de JAK parecen asociarse a una cinética de respuesta relativamente rápida en comparación a los anticuerpos monoclonales18. Cabe tener en cuenta, sin embargo, que existe cierta redundancia inmunológica en el sistema JAK-STAT: hay más de 50 citoquinas y solo 4 isoformas de JAK. Existe, por tanto, una convergencia de mecanismos inmunológicos y, por ello, su inhibición se asocia a una menor especificidad19. En la estrategia de inhibición de las JAK se emplean moléculas pequeñas, de administración vía oral y tópica, que permiten interferir en la respuesta de citoquinas implicadas en la DA de una forma más amplia, aunque intermitente, y que también será dependiente de la selectividad del fármaco, es decir, de la inhibición más o menos predominante de alguna de las JAK16,20.
Inhibidores de JAK de administración oralEn este apartado se revisan los iJAK sistémicos, recientemente autorizados o en vías de aprobación para el tratamiento de la DA moderada-grave (tabla 2).
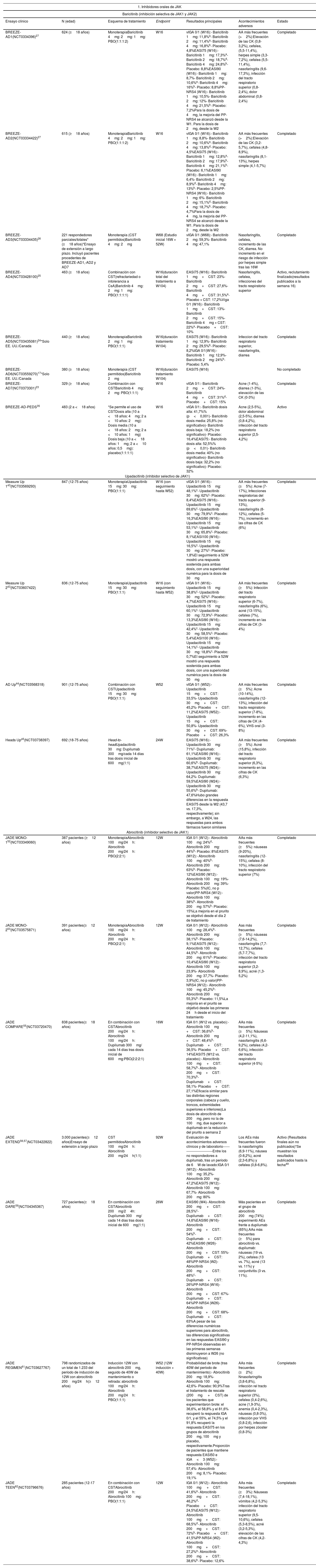
Resultados de los principales ensayos clínicos de los inhibidores de JAK en dermatitis atópica
| 1. Inhibidores orales de JAK | ||||||
|---|---|---|---|---|---|---|
| Baricitinib (inhibición selectiva de JAK1 y JAK2) | ||||||
| Ensayo clínico | N (edad) | Esquema de tratamiento | Endpoint | Resultados principales | Acontecimientos adversos | Estado |
| BREEZE-AD1(NCT0334396)27 | 624 (≥18 años) | MonoterapiaBaricitinib 4mg: 2mg: 1mg: PBO(1:1:1:2) | W16 | vIGA 0/1 (W16):- Baricitinib 1mg: 11,8%a- Baricitinib 2mg: 11,4%a- Baricitinib 4mg: 16,8%b- Placebo: 4,8%EASI75 (W16):- Baricitinib 1mg: 17,3%a- Baricitinib 2mg: 18,7%d- Baricitinib 4mg: 24,8%b- Placebo: 8,8%EASI90 (W16):- Baricitinib 1mg: 8,7%- Baricitinib 2mg: 10,6%a- Baricitinib 4mg: 16%b- Placebo: 8,8%PP-NRS4 (W16):- Baricitinib 1mg: 10,5%- Baricitinib 2mg: 12%- Baricitinib 4mg: 21,5%b- Placebo: 7,2%Para la dosis de 4mg, la mejoría del PP-NRS4 se alcanzó desde la W1. Para la dosis de 2mg, desde la W2 | AA más frecuentes (>2%):Elevación de las CK (0,8-3,2%), cefalea, (5,5-11,4%), herpes simple (3,3-7,2%), cefalea (5,5-11,4%), nasofaringitis (9,6-17,3%), infección del tracto respiratorio superior (0,8-2,4%), dolor abdominal (0,8-2,4%) | Completado |
| BREEZE-AD2(NCT03334422)27 | 615 (≥18 años) | MonoterapiaBaricitinib 4mg: 2mg: 1mg: PBO(1:1:1:2) | W16 | vIGA 0/1 (W16):- Baricitinib 1mg: 8,8%- Baricitinib 2mg: 10,6%a- Baricitinib 4mg: 13,8%d- Placebo: 4,5%EASI75 (W16):- Baricitinib 1mg: 12,8%a- Baricitinib 2mg: 17,9%b- Baricitinib 4mg: 21,1%b- Placebo: 6,1%EASI90 (W16):- Baricitinib 1mg: 6,4%- Baricitinib 2mg: 8,9%d- Baricitinib 4mg: 13%b- Placebo: 2,5%PP-NRS4 (W16):- Baricitinib 1mg: 6%- Baricitinib 2mg: 15,1%d- Baricitinib 4mg: 18,7%b- Placebo: 4,7%Para la dosis de 4mg, la mejoría del PP-NRS4 se alcanzó desde la W1. Para la dosis de 2mg, desde la W2 | AA más frecuentes (>2%):Elevación de las CK (3,2-5,7%), cefalea (4,8-8,9%), nasofaringitis (8,1-13%), herpes simple (4,1-5,7%) | Completado |
| BREEZE-AD3(NCT03334435)26 | 221 respondedores parciales/totales* (≥18 años)*Ensayo de extensión a largo plazo. Incluyó pacientes procedentes de BREEZE-AD1,-AD2 y AD7 | Monoterapia (CST permitidos)Baricitinib 4mg: 2mg | W68 (Estudio inicial 16W + 52W) | vIGA 0/1 (W68):- Baricitinib 2mg: 59,3%- Baricitinib 4mg: 47,1% | Nasofaringitis, cefalea, incremento de las CK, diarrea. No incremento en el riesgo de infección por herpes simple tras las 16W | Completado |
| BREEZE-AD4(NCT03428100)25 | 463 (≥18 años) | Combinación con CST(refractariedad o intolerancia a CsA)Barictinib 4mg: 2mg: 1mg: PBO(1:1:1:1) | W16(duración total del tratamiento a W104) | EASI75 (W16):- Baricitinib 1mg+CST: 23%- Baricitinib 2mg+CST: 27,6%- Baricitinib 4mg+CST: 31,5%a- Placebo + CST: 17,2%Viga 0/1 (W16):- Baricitinib 1mg+CST: 13%- Baricitinib 2mg+CST: 15%- Baricitinib 4mg + CST: 22%a- Placebo+CST: 10% | Nasofaringitis, cefalea, infecciones del tracto respiratorio superior | Activo, reclutamiento finalizado(resultados publicados a la semana 16) |
| BREEZE-AD5(NCT03435081)24*Solo EE. UU./Canada | 440 (≥18 años) | MonoterapiaBarictinib 2mg: 1mg: PBO(1:1:1) | W16(duración tratamiento W104) | EASI75 (W16):- Baricitinib 1mg: 12,9%- Baricitinib 2mg: 29,5%b- Placebo: 8,2%IGA 0/1(W16):- Baricitinib 1mg: 12,9%- Baricitinib 2mg: 24%b- Placebo: 5,4% | Infeccion del tracto respiratorio superior, nasofaringitis, diarrea | Completado |
| BREEZE-AD6(NCT03559270)17*Solo EE. UU./Canada | 380 (≥18 años) | Monoterapia (CST permitidos)Baricitinib 2mg | W16(duración tratamiento W104) | EASI75 (W16) | No completado | |
| BREEZE-AD7(NCT0373301)23 | 329 (≥18 años) | Combinación con CSTBaricitinib 4mg: 2mg: PBO(1:1:1) | W16 | vIGA 0/1:- Baricitinib 2mg+CST: 24%- Baricitinib 4mg+CST: 31%d- Placebo+CST: 15% | Acne (1-4%), diarrea (1-3%), elevación de las CK (0-3%) | Completado |
| BREEZE-AD-PEDS35 | 483 (2 a <18 años) | *Se permite el uso de CSTDosis alta (10 a <18 años: 4mg; 2 a <10 años: 2mg): Dosis media (10 a <18 años: 2mg; 2 a <10 años: 1mg) Dosis baja (10 a <18 años: 1mg; 2 a <10 años: 0,5mg): placebo(1:1:1:1) | W16 | vIGA 0/1:- Baricitinib dosis alta: 41,7%% (p<0,001)- Baricitinib dosis media: 25,8% (no significativo)- Baricitinib dosis baja: 18,2% (no significativo)- Placebo: 16,4%EASI75:- Baricitinib dosis alta: 52,5%% (p<0,01)- Baricitinib dosis media: 40% (no significativo)- Baricitinib dosis baja: 32,2% (no significativo)- Placebo: 32% | Acne (2,5-5%), dolor abdominal (2,5-5%), diarrea (0,8-4,2%), infección del tracto respiratorio superior (2,5-4,2%) | Activo |
| Upadacitinib (inhibidor selectivo de JAK1) | ||||||
| Measure Up 143(NCT03569293) | 847 (12-75 años) | MonoterapiaUpadacitinib 15mg: 30mg: PBO(1:1:1) | W16 (con seguimiento hasta W52) | vIGA 0/1 (W16):- Upadacitinib 15mg: 48,1%c- Upadacitinib 30mg: 62%c- Placebo: 8,4%EASI75 (W16):- Upadacitinib 15mg: 69,6%c- Upadacitinib 30mg: 79,9%c- Placebo: 16,3%EASI90 (W16):- Upadacitinib 15mg: 53,1%c- Upadacitinib 30mg: 65,8%c- Placebo: 8,1%EASI100 (W16):- Upadacitinib 15mg: 16,5%c- Upadacitinib 30mg: 27%c- Placebo: 1,8%El seguimiento a 52W mostró una respuesta sostenida para ambas dosis, con una superioridad numérica para la dosis de 30mg | AA más frecuentes (≥5%): Acne (7-17%), Infecciones respiratorias del tracto superior (9-13%), nasofaringitis (8-12%), cefalea (5-7%), incremento en las cifras de CK (6%) | Completado |
| Measure Up 243(NCT03607422) | 836 (12-75 años) | MonoterapiaUpadacitinib 15mg: 30mg: PBO(1:1:1) | W16 (con seguimiento hasta W52) | vIGA 0/1 (W16):- Upadacitinib 15mg: 38,8%c- Upadacitinib 30mg: 52%c- Placebo: 4,7%EASI75 (W16):- Upadacitinib 15mg: 60,1%c- Upadacitinib 30mg: 72,9%c- Placebo: 13,3%EASI90 (W16):- Upadacitinib 15mg: 42,4%c- Upadacitinib 30mg: 58,5%c- Placebo: 5,4%EASI100 (W16):- Upadacitinib 15mg: 14,1%c- Upadacitinib 30mg: 18,8%c- Placebo: 0,7%El seguimiento a 52W mostró una respuesta sostenida para ambas dosis, con una superioridad numérica para la dosis de 30mg | AA más frecuentes (≥5%): Infección del tracto respiratorio superior (6-7%), nasofaringitis (6%), acné (13-15%), cefalea (7%), incremento en las cifras de CK (3-4%) | Completado |
| AD Up44(NCT03568318) | 901 (12-75 años) | Combinación con CSTUpadacitinib 15mg: 30mg: PBO(1:1:1) | W52 | vIGA 0/1 (W52):- Upadacitinib 15mg+CST: 33,5%- Upadacitinib 30mg+CST: 45,2%- Placebo+CST: 11,2%EASI75 (W52):- Upadacitinib 15mg+CST: 50,8%- Upadacitinib 30mg+CST: 69%- Placebo+CST: 26,3% | AA más frecuentes (≥5%): Acne (10-14%), nasofaringitis (12-13%), infección del tracto respiratorio superior (7-8%), incremento en las cifras de CK (4-6%), VHS oral (3-8%) | Completado |
| Heads Up45(NCT03738397) | 692 (18-75 años) | Head-to-headUpadacitinib 30mg: Dupilumab 300mg/cada 14 días tras dosis inicial de 600mg(1:1) | 24W | EASI75 (W16):- Upadacitinib 30mg: 71%c- Dupilumab: 61,1%EASI90 (W16):- Upadacitinib 30mg: 60,6%b- Dupilumab: 38,7%EASI75 (W24):- Upadacitinib 30mg: 64,2%- Dupilumab: 59,5%EASI90 (W24):- Upadacitinib 30mg: 55,6%b- Dupilumab: 47,6%Hubo grandes diferencias en la respuesta EASI75 desde la W2 (43,7 vs. 17,3%, respectivamente); sin embargo, a W24, las respuestas para ambos fármacos fueron similares | AA más frecuentes (≥5%): Acné (15,8%), infección del tracto respiratorio superior (6,3%), incremento en las cifras de CK (6,3%) | Completado |
| Abrocitinib (inhibidor selectivo de JAK1) | ||||||
| JADE MONO-152(NCT03349060) | 387 pacientes (≥12 años) | MonoterapiaAbrocitinib 100mg/24h: Abrocitinib 200mg/24h: PBO(2:2:1) | 12W | IGA 0/1 (W12):- Abrocitinib 100mg: 24%d- Abrocitinib 200mg: 44%b- Placebo: 8%EASI75 (W12):- Abrocitinib 100mg: 40%b- Abrocitinib 200mg: 63%b- Placebo: 12%EASI90 (W12):- Abrocitinib 100mg: 19%- Abrocitinib 200mg: 39%- Placebo: 5%(IC, no p valor)PP-NRS4 (W12):- Abrocitinib 100mg: 38%b- Abrocitinib 200mg: 57%b- Placebo: 15%La mejoría en el prurito se objetivó desde el día 2 de tratamiento | AAs más frecuentes (≥5%): náuseas (9-20%), nasofaringitis (12-15%), cefalea (8-10%), infección del tracto respiratorio superior (7%) | Completado |
| JADE MONO-253(NCT03575871) | 391 pacientes(≥12 años) | MonoterapiaAbrocitinib 100mg/24h: Abrocitinib 200mg/24h: PBO(2:2:1) | 12W | IGA 0/1 (W12):- Abrocitinib 100mg: 28,4%b- Abrocitinib 200mg: 38,1%b- Placebo: 9,1%EASI75 (W12):- Abrocitinib 100mg: 44,5%b- Abrocitinib 200mg: 61%b- Placebo: 10,4%EASI90 (W12):- Abrocitinib 100mg: 23,9%- Abrocitinib 200mg: 37,7%- Placebo: 3,9%(IC, no p valor)PP-NRS4 (W12):- Abrocitinib 100mg: 45,2%b- Abrocitinib 200mg: 55,3%b- Placebo: 11,5%La mejoría en el prurito se objetivó desde las primeras 24h desde el inicio del tratamiento | Aas más frecuentes (≥5%): náuseas (7,6-14,2%), nasofaringitis (7,7-12,7%), cefalea (5,7-7,7%), infección del tracto respiratorio superior (3,2-8,9%), acné (1,3-5,2%) | Completado |
| JADE COMPARE55(NCT03720470) | 838 pacientes(≥18 años) | En combinación con CSTAbrocitinib 200mg/24h: Abrocitinib 100mg/24h: Dupilumab 300mg/ cada 14 días tras dosis inicial de 600mg:PBO(2:2:2:1) | 16W | IGA 0/1 (W12 vs. placebo):- Abrocitinib 100mg +CST: 36,6%b- Abrocitinib 200mg +CST: 48,4%b- Dupilumab+CST: 36,5%- Placebo+CST: 14%EASI75 (W12 vs. placebo):- Abrocitinib 100mg +CST: 58,7%b- Abrocitinib 200mg+CST: 70,3%b- Dupilumab+CST: 58,1%- Placebo+CST: 27,1%Eficacia similar para las distintas regiones corporales (cabeza y cuello, troncos, extremidades superiores e inferiores)La dosis de abrocitinib de 200mg, pero no la de 100mg, due superior a dupilumab en la reducción del prurito a semana 2 | AAs más frecuentes (≥5%): Náuseas (4,2-11,1%), nasofaringitis (6,6-9,2%), cefalea (4,2-6,6%), infección del tracto respiratorio superior (4-5%) | Completado |
| JADE EXTEND56,57(NCT03422822) | 3.000 pacientes(≥12 años)Ensayo de extensión a largo plazo | CST permitidosAbrocitinib 100mg/24h: Abrocitinib 200mg/24h(1:1) | 92W | Evaluación de acontecimientos adversos clínicos y de laboratorio------------------------------Entre los no respondedores a dupilumab, tras un período de 6W de lavado:IGA 0/1 (W12):- Abrocitinib 100mg: 35,2%- Abrocitinib 200mg: 47,2%EASI75 (W12):- Abrocitinib 100mg: 67,7%- Abrocitinib 200mg: 80% | Los AEs más frecuentes fueron la nasofaringitis (6,9-11%), náusea (0-8,2%), acné (2,3-6,8%) y cefalea (0,8-6,8%). | Activo (Resultados finales aún no publicados)*Se muestran los resultados publicados hasta la fecha49 |
| JADE DARE59(NCT04345367) | 727 pacientes(≥18 años) | En combinación con CSTAbrocitinib 200mg/24h: Dupilumab 300mg/ cada 14 días tras dosis inicial de 600mg(1:1) | 26W | EASI90 (W4)- Abrocitinib 200mg +CST: 28,5%c- Dupilumab+CST: 14,6%EASI90 (W16)- Abrocitinib 200mg+CST: 54%b- Dupilumab+CST: 42%EASI90 (W26)- Abrocitinib 200mg+CST: 55%- Dupilumab+CST: 48%PP-NRS4 (W2)- Abrocitinib 200mg+CST: 48%c- Dupilumab+CST: 26%PP-NRS4 (W16)- Abrocitinib 200mg+CST: 67%- Dupilumab+CST: 64%PP-NRS4 (W26)- Abrocitinib 200mg+CST: 68%- Dupilumab+CST: 63%A pesar de las diferencias numéricas superiores para abrocitinib, las diferencias significativas en las respuestas EASI90 y PP-NRS4 observadas en las primeras semanas disminuyeron a W26 (no significativas) | Más pacientes en el grupo de abrocitinib 200mg (74%) experimentó AEs frente a dupilumab (65%).AAs más frecuentes (≥5%) para abrocitinib vs. dupilumab: náuseas (19 vs. 2%), cefalea (13 vs. 7%), acné (13 vs. 11%) y conjuntivitis (3 vs. 11%). | Completado |
| JADE REGIMEN61(NCT03627767) | 798 randomizados de un total de 1.233 del período de inducción de 12W con abrocitinib 200mg/24h(≥12 años) | Inducción 12W con abrocitinib 200mg, seguido de 40W de mantenimiento o retirada: abrocitinib 100mg/24h: Abrocitinib 200mg/24h: PBO(1:1:1) | W52 (12W inducción + 40W) | Probabilidad de brote (tras 40W del período de mantenimiento):- Abrocitinib 200mg: 18,9%- Abrocitinib 100mg: 42,6%- Placebo: 90,9%Tras el tratamiento de rescate (200mg + CST) de los pacientes que experimentaron brote: el 36,6%, el 58,8% y el 81,6% recuperó la respuesta IGA 0/1, y el 55%, el 74,5% y el 91,8% recuperó la respuesta EASI75 en los grupos de abrocitinib 200mg, 100mg y placebo, respectivamente.Proporción de pacientes que mantiene respuesta EASI50 e IGA<3 (W52):- Abrocitinib 100mg: 57,4%- Abrocitinib 200mg: 8,1%- Placebo: 19,1% | AAs más frecuentes (≥2%): Nnasofaringitis (3,8-6,8%), infección rel tracto respiratorio superior (3%), cefalea (0,4-2,6%), acne (1,9-3%), anemia (0,4-2,3%), náuseas (0,8-3%), infección por VHS (0,8-2,6), infección por herpes zóoster (0,8-3%) | Completado |
| JADE TEEN62(NCT03796676) | 285 pacientes (12-17 años) | En combinación con CSTAbrocitinib 200mg/24h: Abrocitinib 100mg: PBO(1:1:1) | 12W | IGA 0/1 (W12):- Abrocitinib 100mg +CST: 41,6%a- Abrocitinib 200mg+CST: 46,2%d- Placebo+CST: 24,5%EASI75 (W12):- Abrocitinib 100mg+CST: 68,5%d- Abrocitinib 200mg+CST: 72%b- Placebo+CST: 41,5%PP-NRS4 (W2)- Abrocitinib 100mg+CST: 27,2%a- Abrocitinib 200mg+CST: 38,6%b- Placebo: 12,6% | AAs más frecuentes (≥3%): Náuseas (7,4-18,1%), vómitos (4,2-5,3%) infección del tracto respiratorio superior (9,5-10,6%), cefalea (5,3-8,5%), acné (3,2-5,3%), elevación de las cifras de CK (4,2-4,3%) | Completado |
| 2. Inhibidores tópicos de JAK | ||||||
|---|---|---|---|---|---|---|
| Ruxolitinib (inhibición selectiva de JAK1 y JAK2) | ||||||
| TRuE-AD172(NCT03745638) | 631 pacientes (≥12 años) | Ruxolitinib crema 0,75%/cada 12h: Ruxolitinib crema 1,5%/cada 12h: vehículo/cada 12h(2:2:1) | W8 | IGA-TS (W8)Ruxolitinib crema 0,75%: 50%Ruxolitinib crema 1,5%: 53,8%Vehículo: 15,1%PP-NRS4 (W8)Ruxolitinib crema 0,75%: 40,4%Ruxolitinib crema 1,5%: 52,2%Vehículo: 15,4% | Sensación de escozor o quemazón en el lugar de aplicación (<1%). Elevación transitoria leve en las cifras de plaquetas a semana 2, manteniéndose en rango normal | Completado |
| TRuE-AD272(NCT03745651) | 618 pacientes (≥12 años) | Ruxolitinib crema 0,75%/cada 12h: Ruxolitinib crema 1,5%/cada 12h: vehículo/cada 12h(2:2:1) | W8 | IGA-TS (W8)Ruxolitinib crema 0,75%: 39%Ruxolitinib crema 1,5%: 51,3%Vehículo: 7,6%PP-NRS4 (W8)Ruxolitinib crema 0,75%: 42,7%Ruxolitinib crema 1,5%: 50,7%Vehículo: 16,3% | Sensación de escozor o quemazón en el lugar de aplicación (<1%). Elevación transitoria leve en las cifras de plaquetas a semana 2, manteniéndose en rango normal | Completado |
| Delgocitinib (inhibición pan-JAK: JAK1, JAK 2, JAK3 y TYK2) | ||||||
| JapicCTI-17355469 | Pacientes (≥16 años) | Delgocitinib 0,5% crema/cada 12h: vehículo2:1 | W4 | Reducción mEASI (W4)Delgocitinib 0,5% crema: −44,3%Vehículo: 1,7%mEASI50 (W4)Delgocitinib 0,5% crema: 51,9%Vehículo: 11,5%Entre los pacientes que continuaron recibiendo delgocitinib en el estudio de extensión, un 69,3% alcanzó el mEASI50 a W28 | Erupción variceliforme de Kaposi, acné, paroniquia | Completado |
| JapicCTI-18406470 | 137 pacientes(2-15 años) | Delgocitinib 0,25% crema/cada 12h: vehículo2:1 | W4 | Reducción mEASI (W4)Delgocitinib 0,5% crema: −39,3%Vehículo: 10,9%mEASI50 (W4)Delgocitinib 0,5% crema: 50,7%Vehículo: 17,6%Entre los pacientes que continuaron recibiendo delgocitinib en el estudio de extensión, un 73,6% alcanzó el mEASI50 a W56 | Foliculitis en el lugar de aplicación, acné en el lugar de aplicación, molluscum contagiosum, verrugas víricas, impétigo | Completado |
AAs: acontecimientos adversos; CST: corticoesteroides tópicos; CK: Creatine kinase; EASI: Eczema Area and Severity Index; mEASI: EASI modificado: se excluye la región de cabeza y cuello del cálculo del EASI; IGA-treatment success (IGA-TS) definido como un eritema claro sin induración ni papulación (IGA-TS 0) o 1 (definido como un eritema claro con muy escasa induración o papulación); JAK: Janus Kinasa; N: número; PBO: placebo; PP-NRS4: ≥4-point improvement from baseline in the Peak Pruritus Numerical Rating Scale; vIGA: validated Investigator Global Assessment; W: Week.
Baricitinib, autorizado en septiembre de 2020 por la EMA para el tratamiento en adultos de la DA moderada-grave, es un iJAK de primera generación, que bloquea de forma selectiva y reversible JAK1 y JAK2, con menor afinidad para JAK3 y TYK2. Inhibe la transducción de la señal de citoquinas Th2, como la IL-4, IL-5 e IL-13, así como la acción de la IL-22 e IL-3121.
La eficacia de este inhibidor de JAK1/2 se demostró inicialmente en un ensayo clínico (EC) de fase 2 en combinación con corticoides tópicos21. Posteriormente, el programa de desarrollo de fase 3 (BREEZE-AD), incluyó 7 EC en adultos21–27. Los ensayos BREEZE-AD1 y BREEZE-AD2, de diseño idéntico, evaluaron la eficacia en monoterapia de baricitinib a dosis diarias de 1, 2 y 4mg vs. placebo por 16 semanas en 1.239 pacientes. El objetivo principal fue la respuesta Investigator Global Assessment (IGA) 0/1, equivalente al aclaramiento completo o casi completo, con una mejoría de ≥2 puntos respecto al basal. Un mayor porcentaje de los pacientes en los grupos de baricitinib 4 y 2mg alcanzó la respuesta IGA 0/1 en comparación a placebo (tabla 2). Cerca de uno de cada 4 pacientes alcanzó una respuesta EASI75 a las 16 semanas de tratamiento para la dosis de 4mg. Se objetivó una reducción rápida del prurito, significativamente superior para la dosis de 4mg vs. placebo tras solo una semana de tratamiento. Las mejorías en el número de despertares nocturnos, el dolor cutáneo, y los índices de calidad de vida fueron significativas para ambas dosis frente a placebo en la semana 127. BREEZE-AD4 se realizó sobre individuos que habían experimentado fallo o intolerancia a ciclosporina25. Por último, el BREEZE-AD7 se realizó con el objetivo de conocer la eficacia de baricitinib a dosis de 4 y 2mg en combinación con corticoides tópicos. La respuesta EASI75 se alcanzó en un 48% para la dosis de 4mg y del 43% para el grupo de 2mg (no estadísticamente significativa para esta dosis), en comparación con el 23% en el grupo placebo, a semana 16. Para ambas dosis, se produjo una reducción significativa del prurito a las 24 horas después de la primera dosis del fármaco. Recientemente, se han publicado los datos del ensayo a largo plazo de BREEZE-AD3, en el que se observa un mantenimiento de la respuesta a 104 semanas de tratamiento. En este ensayo, los pacientes que habían sido respondedores (IGA 0/1) o respondedores parciales (IGA 2) a baricitinib 4mg/24h, a semana 52 fueron realeatorizados a recibir la misma dosis, 2mg o placebo. Entre los pacientes que mantuvieron baricitinib 4mg, a semana 104 un 47,6% mantuvo la respuesta IGA 0/1 (vs. 51,2% a semana 52) y un 73,8% la respuesta EASI75 (vs. 82,1% a semana 52). Entre los que se descendió la dosis a 2mg/24h una mayoría mantuvieron la respuesta, aunque con cierto descenso en la respuesta IGA 0/1 (35,7%) y la respuesta EASI75 (58,3%)28.
Desde el punto de vista molecular, se ha objetivado en 124 pacientes tratados con baricitinib una disminución gradual de los niveles de IL-19 sérica, un marcador de proliferación de los queratinocitos29. Un estudio sobre 14 pacientes japoneses encontró una asociación positiva entre la mejoría de la DA y el descenso de la IL-22 tras 4 semanas de tratamiento con baricitinib 4mg30.
En cuanto a la seguridad, la mayoría de los acontecimientos adversos en el programa de desarrollo de baricitinib en la DA fueron leves o moderados en gravedad e incluyeron las elevaciones asintomáticas de las CK, la nasofaringitis, la cefalea y la diarrea. Respecto a las alteraciones hematológicas, puede asociarse, a diferencia de otros inhibidores de JAK, a un incremento discreto en las cifras de plaquetas, sin haberse asociado a acontecimientos adversos. La infección grave más frecuente fue el eccema herpeticum, con una tasa de incidencia de 1,4 por 100 pacientes/año en el grupo de todos los pacientes expuestos a baricitinib (N=2.636). La incidencia de infección por herpes simple fue superior para la dosis de 4mg (6,1%) comparada con la de 2mg (3,6%) y placebo (2,7%) a las 16W, con una tasa de incidencia global de 6,7 por 100 pacientes/año en todo el grupo expuesto a baricitinib. La tasa de incidencia de Major Adverse Cardiovascular Events (MACE) fue de 0,15 y hubo 3 casos de tromboembolismo pulmonar (tasa de incidencia de 0,06), aunque no se consideraron atribuibles al fármaco dado que la incidencia fue similar a la de la población general y los pacientes presentaban factores de riesgo para dichos eventos31. Recientemente, se ha publicado una comparación de la seguridad de baricitinib en población con artritis reumatoide (AR), alopecia areata y DA que estratifica a los pacientes, además, de acuerdo a factores de riesgo individuales. Los resultados muestran que la incidencia de los acontecimientos adversos de interés, como los MACE, la enfermedad tromboembólica o las neoplasias, son similares en la población de DA y alopecia areata tratada con baricitinib respecto a lo esperable de acuerdo a la literatura, independientemente de la presencia de otros factores de riesgo, como la edad ≥65 años. En cualquier caso, se aconseja individualizar la decisión del tratamiento de acuerdo los factores de riesgo y la carga de la enfermedad del paciente32.
En conjunto, baricitinib a 4mg ha demostrado una mayor magnitud en la respuesta terapéutica y un inicio de acción más rápido vs. placebo que la dosis de 2mg, con un perfil de seguridad aceptable para ambas dosis del fármaco. Por ello, la dosis recomendada33 es de 4mg al día, y se aconseja el uso de 2mg/24h en circunstancias particulares (tabla 3) siendo también posible la optimización del fármaco a esta dosis en un subgrupo de los pacientes tratados34.
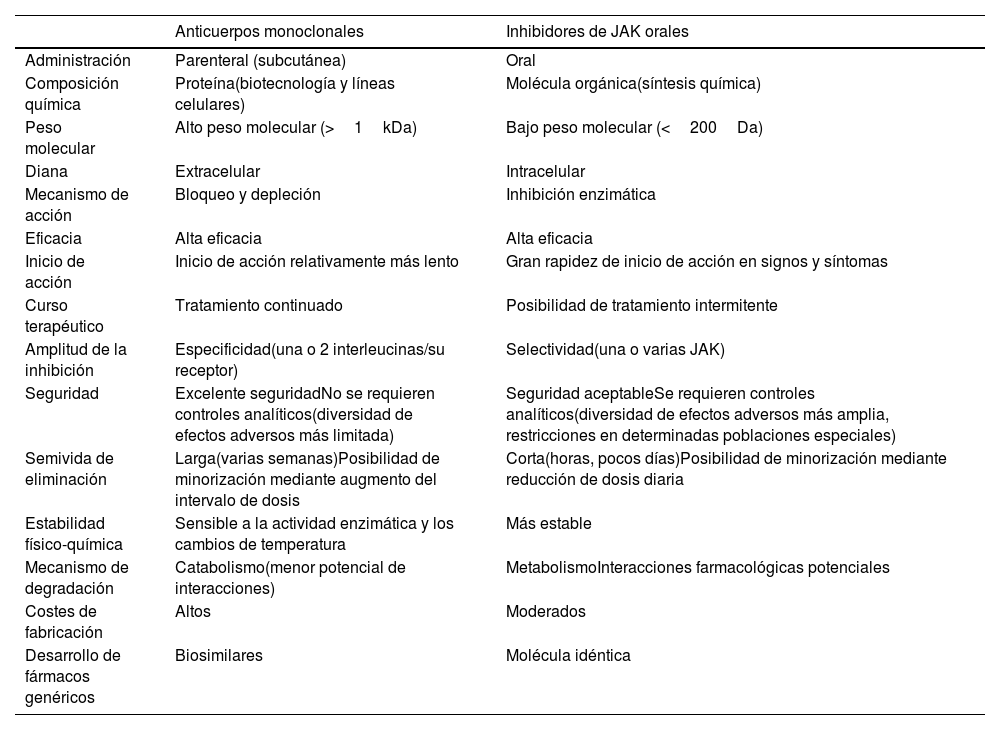
Características generales de los anticuerpos monoclonales y los inhibidores de JAK
| Anticuerpos monoclonales | Inhibidores de JAK orales | |
|---|---|---|
| Administración | Parenteral (subcutánea) | Oral |
| Composición química | Proteína(biotecnología y líneas celulares) | Molécula orgánica(síntesis química) |
| Peso molecular | Alto peso molecular (>1kDa) | Bajo peso molecular (<200Da) |
| Diana | Extracelular | Intracelular |
| Mecanismo de acción | Bloqueo y depleción | Inhibición enzimática |
| Eficacia | Alta eficacia | Alta eficacia |
| Inicio de acción | Inicio de acción relativamente más lento | Gran rapidez de inicio de acción en signos y síntomas |
| Curso terapéutico | Tratamiento continuado | Posibilidad de tratamiento intermitente |
| Amplitud de la inhibición | Especificidad(una o 2 interleucinas/su receptor) | Selectividad(una o varias JAK) |
| Seguridad | Excelente seguridadNo se requieren controles analíticos(diversidad de efectos adversos más limitada) | Seguridad aceptableSe requieren controles analíticos(diversidad de efectos adversos más amplia, restricciones en determinadas poblaciones especiales) |
| Semivida de eliminación | Larga(varias semanas)Posibilidad de minorización mediante augmento del intervalo de dosis | Corta(horas, pocos días)Posibilidad de minorización mediante reducción de dosis diaria |
| Estabilidad físico-química | Sensible a la actividad enzimática y los cambios de temperatura | Más estable |
| Mecanismo de degradación | Catabolismo(menor potencial de interacciones) | MetabolismoInteracciones farmacológicas potenciales |
| Costes de fabricación | Altos | Moderados |
| Desarrollo de fármacos genéricos | Biosimilares | Molécula idéntica |
JAK: Janus Kinasa.
Actualmente se encuentra en marcha un EC fase 3 en población pediátrica de 2 a 17 años de edad (NCT03952559) cuyos primeros resultados demuestran un mayor beneficio para la dosis más alta de baricitinib (4mg en pacientes de 10-17 años y 2mg para los pacientes de 2-9 años de edad), alcanzándose la respuesta IGA 0/1 en el 41,7% y la respuesta EASI75 en el 52,5% a W1635.
Recientemente, se han publicado los primeros datos en vida real de baricitinib en la población adulta, aunque aún limitados en número de pacientes, tiempo de tratamiento y evaluación de la optimización de dosis36–39. En una serie japonesa de 14 pacientes tratados con baricitinib, el 64% alcanzó la respuesta EASI75 y el 36% el EASI90 a W12. Otro estudio japonés (N=36), describe una mayor probabilidad de respuesta en las extremidades inferiores respecto a la localización de cabeza y cuello a W1240. Una serie alemana, con 12 pacientes tratados, con una respuesta EASI75 en el 90% de los pacientes a W1238. El registro Bioday, de Países Bajos, incluye 51 pacientes tratados y reportan una posibilidad de respuesta IGA 0/1 del 22%, dato similar al de los ensayos clínicos. En un tercio de los pacientes se suspendió el tratamiento por falta de eficacia39. Considerando la indicación de baricitinib en alopecia areata, existen casos descritos en la literatura del tratamiento de pacientes con DA y AA concomitante41.
UpadacitinibUpadacitinib inhibe selectivamente JAK1. El fármaco fue aprobado por la EMA para el tratamiento de la DA moderada-grave en ≥12 años de edad en agosto de 202142, y su comercialización en España fue autorizada en abril de 2022.
Dos EC de fase 3 de diseño idéntico evaluaron la eficacia de upadacitinib 15 y 30mg al día en monoterapia comparado con placebo en pacientes con DA moderada-grave de 12 a 75 años de edad (Measure Up1 y Measure Up 2)43. Para ambas dosis, se alcanzaron los enpoints principales (vIGA 0/1 y EASI75) a la semana 16, tanto en Measure Up 1 (vIGA 0/1: upadacitinib 15mg/48,1%, upadacitinib 30mg/62%, vs. placebo 8,4% [p<0,0001]; respuesta EASI75: upadacitinib 15mg/69,6%, upadacitinib 30mg/79,9%, vs. placebo 16,3% [p<0,0001]) como en Measure Up 2 (vIGA 0/1: upadacitinib 15mg/38,8%, upadacitinib 30mg/52%, vs. placebo 4,7% [p<0,0001]; respuesta EASI75: upadacitinib 15mg/60,1%, upadacitinib 30mg/72,9%, vs. placebo 13,3% [p<0,0001]). También se alcanzaron los objetivos secundarios para ambas dosis. La incidencia de acontecimientos adversos (AAs) graves y aquellos que motivaron la salida del estudio fueron similares en los 3 grupos. La incidencia de infección por herpes zóster fue superior en los grupos de upadacitinib 15 y 30mg frente a placebo (20 [1,8%] sobre el total de 1.124 pacientes con upadacitinib, vs. 2 casos [<1%] de 559 en los grupos placebo); en ningún caso motivó la discontinuación del fármaco43. Otro EC fase 3 a 52 semanas (AD Up), evaluó la eficacia y seguridad de upadacitinib en combinación con corticoides tópicos. De nuevo, se alcanzaron los objetivos de respuesta principales (vIGA 0/1 y EASI75) para ambas dosis, no solo en términos de eficacia si no también en la rapidez para el inicio de acción, alcanzando diferencias significativas desde la semana 2 de tratamiento, con una respuesta sostenida en la semana 52, sin nuevos hallazgos en términos de seguridad en dicho período de seguimiento44,45.
Por último, el ensayo Heads Up46, comparó upadactinib 30mg vs. dupilumab a 24 semanas, en este caso únicamente en adultos. A las 16 semanas, un 71% de los tratados con upadacitinib 30mg/24h alcanzó la respuesta EASI75, comparado con el 61,1% en el grupo de dupilumab (p=0,006). Updacitinib demostró un inicio de acción más rápido, con un 43,7% alcanzando el EASI75 en la semana 2 en el grupo de upadacitinib, comparado con el 17,4% del grupo de dupilumab. A semana 16, la proporción de pacientes que alcanzó la respuesta EASI90 (60,6% en el grupo de upadactinib y 38,7% en el grupo de dupilumab) y EASI100 (23,2 y 7,6%, respectivamente) fue superior con upadacitinib (p<0,001). La respuesta, sin embargo, a W24 fue similar para ambos fármacos. Desde el punto de vista de la seguridad, hubo una mayor incidencia de eccema herpeticum, herpes zóster, y alteraciones de laboratorio en el grupo de upadacitinib, mientras que con dupilumab se reportaron más casos de conjuntivitis y reacciones en el punto de inyección. Se produjo una defunción en el grupo de upadacitinib debido a bronconeumonía por influenza.
Existe un número creciente de publicaciones en vida real sobre el uso de upadacitinib en la DA, incluyendo adultos y población adolescente ≥12 años de edad, con unas perspectivas de respuesta similares a las observadas en los EC, y destaca como efecto adverso la posible aparición de acné47–49. Por otro lado, se ha descrito la resolución de la conjuntivitis inducida por dupilumab tras el cambio a upadacitinib, con buen control de la DA50.
AbrocitinibAbrocitinib es un inhibidor selectivo de JAK1 aprobado por la EMA en diciembre del 2021 para el tratamiento de la DA moderada-grave en adultos51. Su eficacia en monoterapia (100 y 200mg/cada 24h) frente a placebo ha sido evaluada en 2 EC de diseño idéntico a 12 semanas (JADE MONO-152 y JADE MONO-253) en pacientes adultos y adolescentes. Para ambas dosis, la respuesta EASI75 e IGA 0/1 fue superior frente a placebo (tabla 2). Respecto a los datos de seguridad, los AAs fueron más frecuentes en el grupo de abrocitinib, pero no hubo diferencias respecto los AAs graves. Los AAs más frecuentes con abrocitinib fueron las náuseas, la cefalea, la nasofaringitis52,53, y la aparición de acné, que fue dosis-dependiente53.
El estudio JADE COMPARE a 16 semanas evaluó la eficacia y la seguridad de abrocitinib (a dosis de 100 y 200mg) frente a dupilumab a dosis estándar y placebo. Todos los pacientes recibieron corticoides tópicos de forma concomitante. El 70,3% de los pacientes en el grupo de abrocitinib 200mg y el 58,7% a dosis de 100mg alcanzaron la respuesta EASI75, en comparación al grupo de dupilumab (58,1%) y placebo (27,1%). No hubo diferencias estadísticamente significativas respecto a los objetivos secundarios para abrocitinib vs. dupilumab, salvo en la mejoría del prurito a la semana 2 para abrocitinib 200mg54-55. Los pacientes de los 2 EC en monoterapia y de JADE COMPARE podían continuar en un ensayo de extensión a largo plazo (JADE EXTEND) de abrocitinib a dosis de 100 y 200mg al día, con posibilidad de uso concomitante de corticoides tópicos, a 92 semanas. Los resultados finales de este ensayo están aún pendientes de publicación56, aunque se han publicado los primeros resultados: los pacientes procedentes de JADE COMPARE que habían recibido dupilumab hasta W14 recibieron placebo oral hasta W20 y posteriormente entraron en JADE EXTEND, siendo realeatorizados a recibir abrocitinib 200 o 100mg al día. Entre los pacientes que no habían sido respondedores a dupilumab, la respuesta EASI75 se alcanzó a W12 en un 80% en los tratados con abrocitinib 200mg, y en un 67,7% para la dosis de 100mg57. Debe considerarse, sin embargo, que tras las 6 semanas de lavado con placebo (W14 a W20), aún podía existir cierto efecto residual de dupilumab58.
El estudio JADE DARE59 ha comparado abrocitinib 200mg vs. dupilumab, asociados a corticoides tópicos. Aunque abrocitinib 200mg fue superior a dupilumab para la respuesta EASI90 a W4 (28,5 vs. 14,6%) y la respuesta PP-NRS4 a W2 (48 vs. 26%), las diferencias entre ambos grupos disminuyeron a lo largo de las 26 semanas, de forma similar a lo evidenciado en el ensayo Heads Up con upadacitinib60.
El estudio JADE REGIMEN61, a 52 semanas, consistió en un período de inducción de 12 semanas en abierto, con abrocitinib 200mg/24h, en pacientes ≥12 años. Los pacientes que alcanzaron la respuesta EASI75 e IGA 0/1 (64,7%) fueron randomizados a recibir abrocitinib 100mg, abrocitinib 200mg o placebo durante 40 semanas. Se evaluó la proporción de pacientes que no presentaba una pérdida de respuesta EASI50 y mantenía un IGA<3, siendo superior en el grupo de abrocitinib 200mg (81,1%) vs abrocitinib 100mg (57,4%) y placebo (19,1%), de modo que ambas dosis mantuvieron la respuesta de forma superior a placebo. Entre los que perdían respuesta, el tratamiento de rescate (abrocitinib 200mg/24h+corticoides tópicos) permitió recuperar la respuesta IGA 0/1 y EASI75 en una mayoría de los pacientes. Por último, el ensayo JADE TEEN62,63, evaluó la eficacia de abrocitinib en combinación con +corticoides tópicos en adolescentes de 12 a 17 años, con datos de eficacia y seguridad similares para este grupo de edad (tabla 2).
Finalmente, en un análisis de seguridad integrado del programa de ensayos clínicos JADE, se incluyeron 3.582 pacientes tratados con abrocitinib, con una exposición de 4.313,4 personas/año. La incidencia de SAE (severe adverse event) fue superior en pacientes ≥65 años en comparación a los más jóvenes. Las infecciones graves fueron el SAEs más frecuente, con una tasa de incidencia de 2,46 (1,79-3,31) para abrocitinib 200mg y de 2,43 (1,66-3,44) para abrocitinib 100mg. Entre las más frecuentes se incluyeron la infección por herpes zóster, herpes simple y neumonía64. En conjunto, abrocitinib 200mg es más efectivo en el tratamiento de la DA, aunque con una incidencia de efectos adversos leves y moderados superior a la observada para dosis de 100mg/24h65.
Otros inhibidores orales de JAKGusacitinib (ASN002)Gusacitinib es el primer inhibidor oral dual JAK/SYK: además de inhibir los cuatro tipos de JAK, inhibe la Spleen tyrosine Kinase (SYK), que regula la diferenciación de los linfocitos B y las células dendríticas, así como la señalización Th17/IL-17. La inhibición de SYK permite controlar la señalización Th17 y mejora la diferenciación terminal de los queratinocitos, lo que podría tener un beneficio adicional en determinados endotipos de DA, añadido al que ofrece la inhibición de JAK (controlando la señalización Th1, Th2 y Th22)66. Un EC fase 1b comparó gusacitinib a dosis diarias de 20, 40 y 80mg vs. placebo por 4 semanas. Una mayor proporción de pacientes en los grupos de 40 y 80mg alcanzó una respuesta EASI50 vs. placebo (100% para el grupo de 40mg y 83% en el grupo de 80mg, vs. 22% en el grupo placebo)67. Un EC fase 2b (NCT03531957)66 no encontró diferencias en la reducción absoluta del EASI respecto a placebo, y el estudio de extensión no se completó finalmente. No hay nuevas investigaciones con gusacitinib en DA; sin embargo, sigue evaluándose su posible indicación en eccema crónico de manos66.
TofacitinibTofacitinib es un inhibidor de JAK1 y JAK3 estudiado a dosis de 5mg/12h y 5mg/24 h vía oral únicamente en una prueba de concepto con 6 pacientes adultos con DA, con una reducción en el SCORAD del 66,6% a las 8-29 semanas de tratamiento respecto al basal. Actualmente no parece que vaya a evaluarse su indicación en DA68.
Inhibidores de JAK de administración tópicaLa terapia tópica es un pilar esencial en el tratamiento de la DA. Actualmente se están investigando algunos iJAK de administración tópica, que podrían conseguir un beneficio local, sin asociarse a los potenciales adversos sistémicos relacionados con los iJAK de administración oral.
DelgocitinibDelgocitinib es un iJAK de primera generación, con un perfil de inhibición pan-JAK (JAK1, JAK2, JAK3 y TYK2). Disponemos de los resultados de un EC fase 3, controlado con vehículo, que evaluó la eficacia y seguridad de delgocitinib al 0,5% en 158 pacientes de ≥16 años. De estos, 106 aplicaron delgocitinib y 52 el vehículo, 2 veces al día, durante 4 semanas. El objetivo principal fue la reducción en el EASI modificado o mEASI (se excluye la región de cabeza y cuello del cálculo del EASI). La reducción en el mEASI fue significativamente superior para delgocitinib vs. vehículo (−44,3 vs. 1,7%, respectivamente, p<0,001). Además, un 51,9% alcanzó una reducción de al menos un 50% respecto al mEASI basal (respuesta mEASI50) vs. el 11% en el grupo placebo. La mejoría del prurito ocurrió tras el primer día de tratamiento. El EC continuó con una segunda etapa de extensión de 24 semanas en abierto, alcanzando la respuesta mEASI50 el 95% de los pacientes y mEASI75 un 49%, con un perfil de seguridad aceptable. Hubo 3 casos de erupción variceliforme de Kaposi relacionados con delgocitinib. No se reportaron linfopenias69. Existe también un EC fase 3 en edad pediátrica (≥2 y ≤15 años) a concentraciones del 0,25 y 0,5%, con resultados en la misma dirección y sin nuevas alertas de seguridad (tabla 2). Como limitaciones, la población de estudio en estos EC era únicamente japonesa70. Delgocitinib en crema al 0,5% está aprobado en Japón desde enero de 2020 para el tratamiento de la DA71. Fuera de Japón, existen en marcha estudios evaluando la eficacia y seguridad de delgocitinib a distintas concentraciones (1, 3, 8 y 20mg/g) en adultos (NCT03725722), así como en población pediátrica a partir de 2 años de edad (NCT03826901) con DA, con una superficie corporal afectada <50%. Por otro lado, se está evaluando su potencial beneficio en el tratamiento del eccema crónico de manos (NCT04871711, NCT04872101).
RuxolitinibRuxolitinib es un inhibidor selectivo de JAK1 y JAK2. Dos EC de fase 3 (TRuE-AD1 y TRuE-AD2) evaluaron la eficacia de ruxolitinib en un total de 1.249 pacientes ≥12 años con DA, a concentraciones del 0,75 y del 1,5% vs. vehículo, aplicado cada 12h, durante 8 semanas (tabla 2). Alrededor del 50% de los pacientes tratados con ruxolitinib alcanzó el objetivo principal de evaluación IGA-treatment success (IGA-TS) 0/1 y la reducción del prurito fue significativa desde las 12h tras la primera aplicación de ruxolitinib 1,5%71. Asimismo, se alcanza un control sostenido a semana 52, con una respuesta IGA-TS 0/1 en un 74,1-77,8% de los pacientes tratados72. En estudios de farmacocinética, la aplicación en una superficie corporal de hasta el 20% no se ha asociado a concentraciones plasmáticas significativas71. En octubre de 2021, ruxolitinib tópico fue aprobado por la FDA para el tratamiento a corto plazo o intermitente de la DA en pacientes ≥12 años de edad, siendo el primer inhibidor tópico de JAK autorizado en DA73. A pesar de que este tratamiento se ha evaluado en monoterapia, podría emplearse en combinación con otros tratamientos tópicos o sistémicos.
BrepocitinibBrepocitinib es un inhibidor tópico de JAK1 y TYK2 que se encuentra en desarrollo para el tratamiento de la DA. Recientemente se han publicado los resultados del EC fase 2b evaluando la eficacia y seguridad de brepocitinib a distintas concentraciones (0,1% cada 24h, 0,3% cada 24 o cada 12h, 1% cada 24 o cada 12h y 3% cada 24h) frente a vehículo cada 24 o cada 12h. A las 6 semanas, los pacientes tratados en los grupos de brepocitinib 1% alcanzaron una reducción del EASI de −70,1% (−82,1 a −58) para la aplicación cada 24h y de −75 (−83,8 a 66,2) para la aplicación cada 12h frente a vehículo (cada 24h: –44,4% [–57,3 a –31,6%]; cada 12h: –47,6% [–57,5 a –37,7%])74.
TofacitinibEn 2016 se publicó un EC de fase 2a de tofacitinib 2% en crema. La aplicación 2 veces al día durante 4 semanas se asoció a una reducción media del EASI del 81,7 vs. el 29,9% en el grupo placebo71. Actualmente no hay más estudios registrados evaluando su desarrollo en esta formulación para la DA.
Cerdulatinib (DMVT-502)Cerdulatinib es un inhibidor dual de JAK y SYK, evaluado en DA únicamente en su administración al 0,37% en gel 2 veces al día un EC fase 1b a 14 días, con reducciones significativas del EASI71. En el momento actual, no hay nuevos EC registrados para su desarrollo en la DA.
Consideraciones de uso en la práctica clínicaElección del tratamientoRecientemente, se han publicado algunas revisiones sistemáticas y metaanálisis, incluyendo los biológicos e iJAK disponibles en la DA, que muestran una alta eficacia para upadacitinib y abrocitinib, superiores a dupilumab en las dosis más altas de ambos iJAK. Asimismo, los iJAK son globalmente seguros en la población con DA, pero presentan potenciales alertas de seguridad, y los efectos adversos también son, en muchas ocasiones, dependientes de la dosis75,76. En este sentido, deberán considerarse distintos aspectos que pueden condicionar la elección del tratamiento en un determinado paciente.
A pesar de las diferencias existentes para cada uno de los nuevos tratamientos disponibles en la DA, pueden establecerse 2 estrategias terapéuticas principales, a través de los 2 grandes grupos de terapias innovadoras sistémicas: el uso de anticuerpos monoclonales o biológicos —más específicos—, o bien la inhibición de JAK —no específica, aunque más o menos selectiva a través de la inhibición de una o varias JAK— (tabla 3).
Los inhibidores de JAK presentan beneficios atendiendo a su inicio de acción globalmente más rápido, que permitiría una respuesta rápida y profunda sobre los síntomas y los signos. Esto podría ser potencialmente relevante en pacientes en los que se precise una rápida mejoría de la DA, por ejemplo, en pacientes con una alta carga de la enfermedad o en una DA eritrodérmica, donde la mejoría rápida permita reducir la posible morbimortalidad asociada. El rápido inicio de acción, así como la breve semivida de eliminación de estos fármacos permite plantear la posibilidad de realizar ciclos de tratamiento, particularmente en pacientes con un curso intermitente o estacional de la DA. Esa breve semivida de eliminación del fármaco también tiene como ventaja la rápida reversibilidad de algunos de sus potenciales efectos adversos al suspenderlo. Asimismo, atendiendo a una inhibición más amplia de citoquinas en comparación a los anticuerpos monoclonales, podría ser útil —desde un punto de vista todavía teórico— en aquellas formas de la DA en las que existe una participación relevante de otras vías no Th2 como, por ejemplo, el eccema atópico psoriasiforme. Por otro lado, debe considerarse la posibilidad de su uso en pacientes que puedan beneficiarse del tratamiento simultáneo de otras enfermedades concomitantes, fundamentalmente otras comorbilidades inflamatorias o autoinmunes no Th2: por ejemplo, baricitinib presenta indicación en alopecia areata, y upadacitinib tiene autorizadas indicaciones en artritis inflamatorias y enfermedad inflamatoria intestinal, y se encuentra en desarrollo su uso en el vitíligo y la hidradenitis supurativa. El uso de un inhibidor de JAK puede ser relevante en pacientes que presenten afectación de la superficie ocular basal grave, dado el potencial empeoramiento de la misma relacionado con los biológicos en la DA. También sería de utilidad en casos en los que se desarrollen efectos adversos persistentes o refractarios, fundamentalmente oculares o cutáneos, con el uso de anticuerpos monoclonales.
En cuanto a los biológicos, dupilumab presenta como ventaja su utilidad e indicación en otras comorbilidades Tipo 2, como el asma. El Consenso Europeo77 en el tratamiento de la DA en poblaciones especiales aconseja priorizar el uso de biológicos sobre los iJAK en pacientes mayores de 65 años, pacientes con historia de cáncer, y en aquellos con infecciones frecuentes o infecciones crónicas como el VHB. Existen publicaciones en vida real acerca de la seguridad favorable de dupilumab en población con enfermedad renal crónica grave y en pacientes trasplantados78, y datos que apuntan a su seguridad en el embarazo y la lactancia79.
La elección del tratamiento vendrá condicionada por las guías o acuerdos particulares de cada comunidad autónoma y centro particular, las preferencias del paciente (por ejemplo, la vía de administración oral vs. subcutánea) y la decisión médica en relación al perfil de paciente, sus riesgos y comorbilidades individuales y las perspectivas de respuesta de acuerdo a la evidencia científica actual. En la tabla 3 se comparan ambos grupos terapéuticos, considerando sus características moleculares principales, con implicaciones en sus ventajas e inconvenientes.
Seguridad de los iJAKLos ensayos con iJAK sistémicos en DA, incluyendo un programa de desarrollo extenso para baricitinib, upadacitinib y abrocitinib, han demostrado un perfil de seguridad globalmente aceptable. La mayor parte de los AAs emergentes fueron leves y transitorios, sin diferencias significativas frente a placebo en el periodo controlado. Desde el punto de vista clínico, es posible la aparición de cefalea y náuseas en alrededor de un 10% de los pacientes, siendo algo superiores para baricitinib y abrocitinib, respectivamente, así como el desarrollo de acné y/o foliculitis, cuya incidencia podría ser superior con upadacitinib35. Los datos en vida real permitirán una mejor caracterización de estos hallazgos. Desde el punto de vista de las infecciones, y condicionado por la inhibición de la respuesta a IFN, destaca la mayor susceptibilidad a las infecciones víricas, habiéndose evidenciado un incremento en las tasas de reactivación de herpes simple —en su mayoría oral— y herpes zóster. No hubo casos de reactivación de tuberculosis ni de infecciones oportunistas. Sin embargo, los iJAK no han sido estudiados en combinación con otros inmunosupresores como ciclosporina o metotrexato, por lo que su uso concomitante no está recomendado en pacientes con DA. Dicha combinación podría incrementar el riesgo de inmunosupresión e infecciones graves80.
Entre los efectos adversos de clase de los iJAK destaca la elevación de las CK, en su mayoría de grado 1, y sin repercusión clínica. También se han detectado elevaciones tanto de LDL como de HDL, sin que se produzca una alteración de la ratio LDL:HDL, por lo que se necesitan datos en el largo plazo para interpretar si estos hallazgos se relacionan o no con posibles AAs cardiovasculares. Respecto al hemograma, los iJAK pueden asociarse a la aparición de neutropenia, linfopenia y anemia, aunque su tasa de incidencia entre los pacientes con DA tratados ha sido muy baja y en su mayoría de grado 1. La administración de baricitinib podría asociarse al desarrollo de trombocitosis, y se han descrito casos de trombocitopenia con abrocitinib. En ambos casos, parece que los niveles presentarían su mayor alteración a la semana 4, y posteriormente se recuperarían hasta los niveles basales durante el tratamiento18,81–83.
Una de las alertas más importantes de seguridad es la relacionada con la enfermedad tromboembólica. En los ensayos publicados se han reportado muy escasos eventos de trombosis venosa profunda (TVP) y tromboembolismo pulmonar (TEP), ocurriendo en pacientes con factores de riesgo preexistentes, como la mayor edad, el tabaquismo, o la inmovilización prolongada. Debido a la baja incidencia global en la población general, es difícil medir la fuerza de la asociación de estos eventos con el uso de los iJAK, por lo que se recomienda considerar los factores de riesgo individuales en la selección del tratamiento. De forma similar, la baja incidencia de neoplasias en los estudios realizados no permite demostrar una mayor aparición respecto a la población general18,81–83.
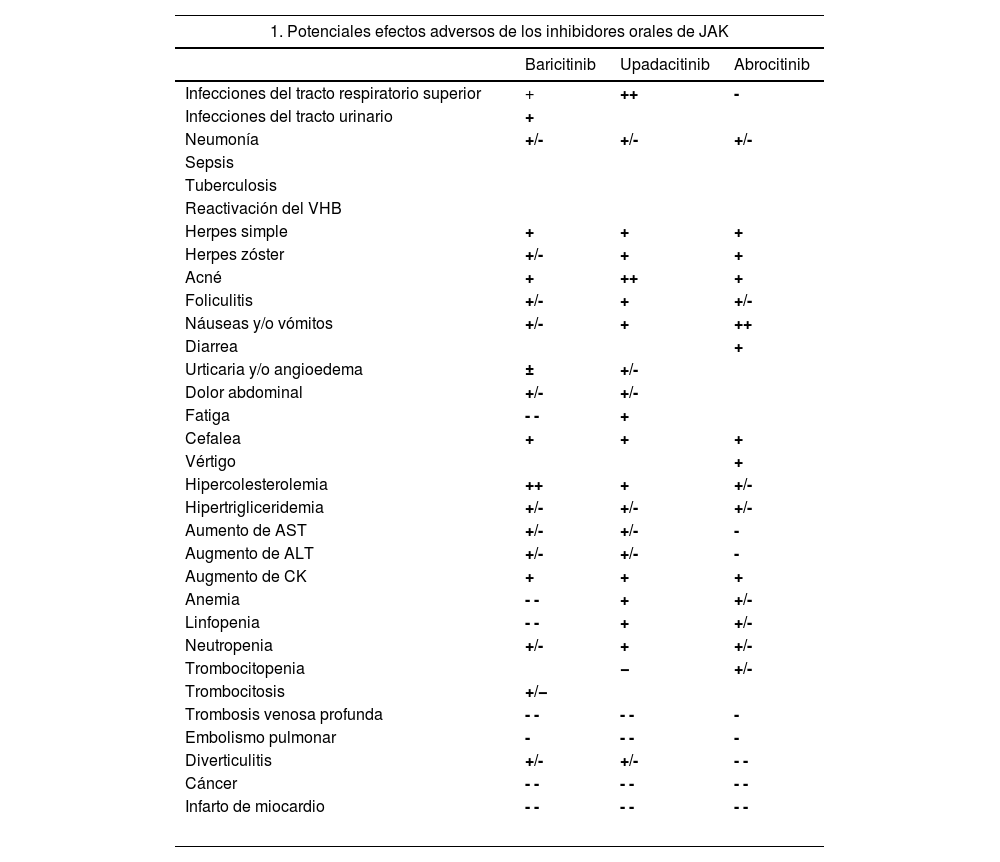
A pesar de la baja incidencia de efectos adversos graves en la población con DA, el 23 de enero de 2023, la EMA publicó unas recomendaciones para reducir su riesgo con el uso de iJAK en enfermedades inflamatorias crónicas. La recomendación establece que debería limitarse el uso de iJAK en los siguientes pacientes, empleándolos solo en aquellos casos en que no se dispongan de otras alternativas disponibles efectivas: pacientes ≥65 años, pacientes con incremento en el riesgo de enfermedad cardiovascular grave como ictus o infarto de miocardio, pacientes fumadores (o exfumadores con un gran consumo acumulado) y pacientes con riesgo de cáncer84. En la tabla 4 se resumen los potenciales efectos adversos frecuentes e infrecuentes relacionados con el uso de iJAK orales, así como las principales recomendaciones de la EMA en la selección del tratamiento18,81–84.
Seguridad de los inhibidores de JAK
| 1. Potenciales efectos adversos de los inhibidores orales de JAK | |||
|---|---|---|---|
| Baricitinib | Upadacitinib | Abrocitinib | |
| Infecciones del tracto respiratorio superior | + | ++ | - |
| Infecciones del tracto urinario | + | ||
| Neumonía | +/- | +/- | +/- |
| Sepsis | |||
| Tuberculosis | |||
| Reactivación del VHB | |||
| Herpes simple | + | + | + |
| Herpes zóster | +/- | + | + |
| Acné | + | ++ | + |
| Foliculitis | +/- | + | +/- |
| Náuseas y/o vómitos | +/- | + | ++ |
| Diarrea | + | ||
| Urticaria y/o angioedema | ± | +/- | |
| Dolor abdominal | +/- | +/- | |
| Fatiga | - - | + | |
| Cefalea | + | + | + |
| Vértigo | + | ||
| Hipercolesterolemia | ++ | + | +/- |
| Hipertrigliceridemia | +/- | +/- | +/- |
| Aumento de AST | +/- | +/- | - |
| Augmento de ALT | +/- | +/- | - |
| Augmento de CK | + | + | + |
| Anemia | - - | + | +/- |
| Linfopenia | - - | + | +/- |
| Neutropenia | +/- | + | +/- |
| Trombocitopenia | − | +/- | |
| Trombocitosis | +/− | ||
| Trombosis venosa profunda | - - | - - | - |
| Embolismo pulmonar | - | - - | - |
| Diverticulitis | +/- | +/- | - - |
| Cáncer | - - | - - | - - |
| Infarto de miocardio | - - | - - | - - |
| 2. Recomendaciones de la EMA para minimizar el riesgo de efectos adversos graves |
|---|
| Se recomienda emplear iJAK sistémicos solo en ausencia de un tratamiento alternativo adecuado en pacientes con uno o más de los siguientes factores:• Edad ≥65 años• Fumadores o exfumadores• Factores de riesgo cardiovasculares adicionales• Factores de riesgo de desarrollo de neoplasias |
| Se recomienda emplear con precaución en pacientes con factores de riesgo de enfermedad tromboembólica:• Antecedentes de enfermedad tromboembólica• Cirugía mayor• Inmovilización prolongada• Anticonceptivos hormonales combinados• Terapia hormonal sustitutiva• Trastorno hereditario de la coagulación• Antecedentes de infarto de miocardio• Insuficiencia cardíaca• Factores de riesgo adicionales: obesidad, diabetes mellitus, hipertensión arterial |
NOTA: Se incluyen los efectos adversos de acuerdo con los detalles contenidos en la ficha técnica de los fármacos. Debe considerarse que se presentan los datos agregados, incluyendo las distintas dosis del fármaco y las potenciales poblaciones clínicas tratadas para cada uno de los fármacos. Las frecuencias específicas dependerán de la dosis empleada y la enfermedad tratada, así como factores individuales del paciente, como la edad. Frecuencia estimada: Muy frecuentes: ++ (≥1/10), frecuentes: + (≥1/100 a <1/10), poco frecuentes: +/- (≥1/1000 a <1/100), raras: - (≥1/10 000 a <1/1000), muy raras: - - (<1/10 000). No reportada.
JAK: Janus Kinasa.
En la tabla 5 se resumen las consideraciones de evaluación previa al tratamiento y durante el seguimiento del mismo, teniendo en cuenta los datos incluidos en la ficha técnica de los iJAK orales aprobados para la DA y la experiencia publicada en la literatura en otras indicaciones, principalmente la artritis reumatoide80,85–87.
Aspectos prácticos de los nuevos inhibidores de JAK
| Evaluación previa al inicio del fármaco |
| 1. Anamnesis y exploración física:• Estimación del riesgo global de enfermedad tromboembólica (antecedentes familiares o personales conocidos, obesidad, tabaquismo, uso de anticonceptivos orales, inmovilización prolongada, edad >45 años, intervenciones quirúrgicas mayores como neurocirugía, cirugías urológicas, ginecológicas o traumatológicas, factores hereditarios —factor V Leiden, mutación 20210 de la protrombina— y trombofilia adquirida —SAF, cáncer—)• Estimación del riesgo cardiovascular global (factores personales de riesgo cardiovascular, uso concomitante de inhibidores de la COX2, prednisona a dosis >7,5mg/24h, anticonceptivos orales)• Exploración dermatológica (evaluación basal en relación a cáncer cutáneo no melanoma)2. Evaluación analítica basal (hemograma y bioquímica):• Hemograma completo (incluyendo fórmula leucocitaria)• Evaluación del perfil hepático (en particular, transaminasas)• Evaluación de la función renal• Se aconseja la evaluación basal del perfil lipídico• No se aconseja la evaluación de las CK de rutina3. Evaluación del riesgo de infección:• Serologías del VHB (anti-HBs, anti-HBc y anti-HBsAg):∘ No aconsejado el uso en pacientes con infección crónica por el VHB∘ En pacientes con exposición previa (anti-HBc+) sin evidencia de replicación activa (HBsAg−), debe evaluarse el ADN-VHB∘ En paciente con anti-HBc+, HBsAG− y con ADN−, puede iniciarse un iJAK evaluando riesgos y beneficios, y con control estrecho junto a hepatología• Serologías del VHC:∘ Debe evaluarse el ARN-VHC. Si es positivo, el paciente debe recibir tratamiento completo frente al VHC antes de iniciar un iJAKSe recomienda solicitar serologías del VIH• Se recomienda el screening de TBC. El riesgo de reactivación de TBC con los iJAK podría ser similar al de los anti-TNF• Radiografía de tórax• Test de tuberculina y/o test de liberación de interferón-γ (IGRA)• En caso de precisar tratamiento, no debe iniciarse un iJAK hasta haber completado 1-2 meses de tratamiento antituberculoso• La respuesta terapéutica de upadacitinib puede disminuir en caso de coadministración con rifampicina (inductor de CYP 3A4)• Serología de varicela-zóster en pacientes sin historia previa de inmunización• Otras serologías: sarampión, rubéola y parotiditis (estado vacunal)4. Evaluación del estado vacunal:• Deben seguirse las recomendaciones nacionales y regionales respecto a la vacunación• Deben recogerse los antecedentes de varicela o infección por herpes zóster, así como el antecedente de inmunización previa:∘ Los factores de riesgo para reactivación del VVZ incluyen la edad >50 años, el sexo femenino, el uso concomitante de corticoides, la infección y la hospitalización∘ Actualmente se aconseja la vacunación previa mediante la vacuna frente al herpes zóster recombinante (Shingrix®, 2 dosis administradas con 2 meses de diferencia) en mayores de 18 en tratamiento con inhibidores de JAK. La vacuna recombinante no está contraindicada durante el tratamiento con iJAK, pero estos podrían afectar a la inmunogenicidad de la misma∘ Las vacunas de virus vivos y virus vivos atenuados están contraindicadas en pacientes en tratamiento con iJAK5. Embarazo y lactancia. El uso de iJAK está contraindicado durante el embarazo y la lactancia. Las mujeres en tratamiento con inhibidores de JAK deben emplear métodos anticonceptivos adecuados:• Baricitinib: durante el tratamiento y al menos durante una semana después de su suspensión• Upadacitinib: durante el tratamiento y al menos durante 4 semanas después de su suspensión• Abrocitinib: durante el tratamiento y al menos durante 4 semanas después de su suspensión• No es necesaria la suspensión preconcepcional del tratamiento con inhibidores de JAK en varones. No se ha demostrado efectos sobre la espermatogénesis6. Evaluación de la medicación concomitante:• Interacciones farmacológicas:∘ Baricitinib: inhibidores OAT3 --> ↑[baricitinib] (p. ej., probenecid: se aconseja emplear la dosis de 2mg/24h)∘ Upadacitinib:• Inhibidores de CYP3A4 --> ↑[upadacitinib] (p. ej., ketoconazol, itraconazol, posaconazol, voriconazol y claritromicina). Debe evitarse la dosis de 30mg/24h Se deben considerar alternativas a los inhibidores potentes del CYP3A4 cuando se utilicen a largo plazo• Inductores del CYP3A4 --> ↓[upadacitinib] (p. ej., rifampicina y fenitoína)∘ Abrocitinib:• Inhibidores del CYP2C19/CYP2C9: ↑[abrocitinib] (p. ej., fluvoxamina y fluconazol)• Inductores del CYP2C19/CYP2C9: ↓[abrocitinib] (p. ej., rifampicina)• Incremento del riesgo de inmunosupresión en combinación con otros fármacos. Se desaconseja el uso de iJAK con ciclosporina u otros inmunosupresores sistémicos en DA |
| Seguimiento clínico y analítico durante el tratamiento |
| 1. Evaluar el riesgo individual respecto a infecciones (p. ej., exposición profesional, viajes, comorbilidades, fármacos concomitantes, signos o síntomas de infección)2. Se recomienda el control analítico a los 3 meses y posteriormente cada 6 meses. Puede considerarse una primera evaluación a las 4 semanas del tratamiento en pacientes con determinados factores de riesgo:• Hemograma:∘ Niveles de hemoglobina (confirmado en 2 analíticas consecutivas):• Valores ≤8g/dl y/o disminución de los niveles >2g/dl respecto al basal: interrumpir el tratamiento hasta su normalización. La reintroducción debe considerarse individualmente, evaluando riesgo/beneficio• Valores >9g/dl no requiere ajuste de dosis∘ Valores absolutos de neutrófilos (confirmado en 2 analíticas consecutivas):• Valores <500mm3: suspensión del tratamiento• Valores entre 500-1.000mm3: reducción de dosis o interrupción temporal hasta normalización con niveles superiores a 1.000mm3• Valores >1.000mm3: no requiere ajuste de dosis∘ Valores absolutos de linfocitos (en 2 analíticas consecutivas):• Valores <500mm3: suspensión del tratamiento• Valores entre 500-750mm3; reducción de dosis o interrupción temporal hasta normalización con niveles superiores a 750mm3• Valores >750mm3: no requiere ajuste de dosis• Evaluación del perfil hepático (ver ajustes de acuerdo al iJAK)• Evaluación de la función renal (ver ajustes de acuerdo al iJAK)• Seguimiento del perfil lipídico (recomendado al tercer mes de tratamiento): en caso de dislipemia, seguir las guías estándar para su tratamiento. Se ha evaluado la potencial interacción farmacológica de diversas estatinas con los inhibidores de JAK disponibles.• La combinación de estatinas con baricitinib, upadacitinib y abrocitinib no parece tener efectos farmacocinéticos relevantes• No se recomienda solicitar los niveles de CK, salvo que exista clínica que lo indique (p. ej., mialgias)3. Seguimiento del riesgo de infecciones:• La evaluación anual de las serologías para VHB, VHC y VIH solo está indicada en pacientes de riesgo para las mismas• La evaluación periódica (p. ej., anual) de la TBC latente debe valorarse en grupos de riesgo para la misma (p. ej., posibilidad de exposición ocupacional)• Si un paciente presenta herpes zóster durante el tratamiento, debe considerarse la interrupción temporal del iJAK hasta la resolución del episodio4. Evaluar el riesgo de neoplasias: programas de cribado de neoplasias de acuerdo a las recomendaciones nacionales para edad y sexo. Evaluación dermatológica (cáncer cutáneo no melanoma y melanoma)5. Vacunación: las vacunas inactivadas pueden administrarse durante el tratamiento con iJAK, aunque potencialmente podría existir una disminución en su inmunogenicidad |
| Consideraciones particulares de los iJAK oral | |||
|---|---|---|---|
| Baricitinib | Upadacitinib | Abrocitinib | |
| Dosis estándar | ≥18-74 años:4mg/24h≥75 años:2mg/24h | >18-64 años:30mg: si alta carga de la enfermedad o respuesta inadecuada a 15mg. Para el mantenimiento se debe considerar la dosis efectiva más baja≥65 y adolescentes>12 años y >30kg: 15mg/24h*Datos limitados en >75 años: utilizar con precaución | >18-64 años:200mg/24h≥65 años:100mg/24h*Datos limitados en >75 años: utilizar con precaución |
| Ajuste de dosis: insuficiencia renal | FGe>30-60ml/min: 2mg/24hFGe<30ml/min: desaconsejado | FGe>30-60ml/min: no precisa ajuste de dosisFGe 15-30ml/min: 15mg/24hFGe<15ml/min: desaconsejado | FGe>30-60ml/min: 100mg/24hFGe 15-30ml/min: 50-100mg/24hFGe<15ml/min: desaconsejado |
| Ajuste de dosis:Insuficiencia hepática | Child-Pugh A-B: no precisa ajuste de dosisChild-Pugh C: desaconsejado | Child-Pugh A-B: no precisa ajuste de dosisChild-Pugh C: desaconsejado | Child-Pugh A-B: no precisa ajuste de dosisChild-Pugh C: desaconsejado |
JAK: Janus Kinasa.
Los inhibidores de JAK podrían cambiar el paradigma de tratamiento de la DA, teniendo en cuenta sus altos niveles de eficacia, el rápido inicio de acción y la inhibición transversal de citoquinas, de especial interés considerando la variabilidad en fenotipos y endotipos de la enfermedad. Aunque globalmente presentan un perfil de seguridad adecuado, la posibilidad de alteraciones analíticas y de acontecimientos adversos graves, aunque infrecuentes, hacen recomendable la adecuada evaluación de los factores de riesgo individuales del paciente al seleccionar el tratamiento y su monitorización en el tiempo. Serán de gran utilidad los datos en vida real, en términos de eficacia y de seguridad, y evaluando su uso en distintos escenarios, como el tratamiento continuado a largo plazo y en una estrategia más breve o intermitente.
FinanciaciónEste trabajo no ha recibido financiación.
Conflicto de interesesM. Munera-Campos ha recibido honorarios por asesoría científica, presentaciones u otras actividades relacionadas de Abbvie, LEO Pharma, Janssen, Sanofi y Galderma, y ha participado como investigadora principal y subinvestigadora en ensayos clínicos: Lilly, LEO Pharma, Novartis, Janssen, Sanofi, Pfizer, AbbVie, Almirall, UCB y Galderma. J.M. Carrascosa ha participado como investigador principal/subinvestigador y/o recibido honorarios como ponente y/o miembro de comité de expertos o steering Comitee para AbbVie, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, BMS, Boehringer ingelheim, Biogen, UCB.



