

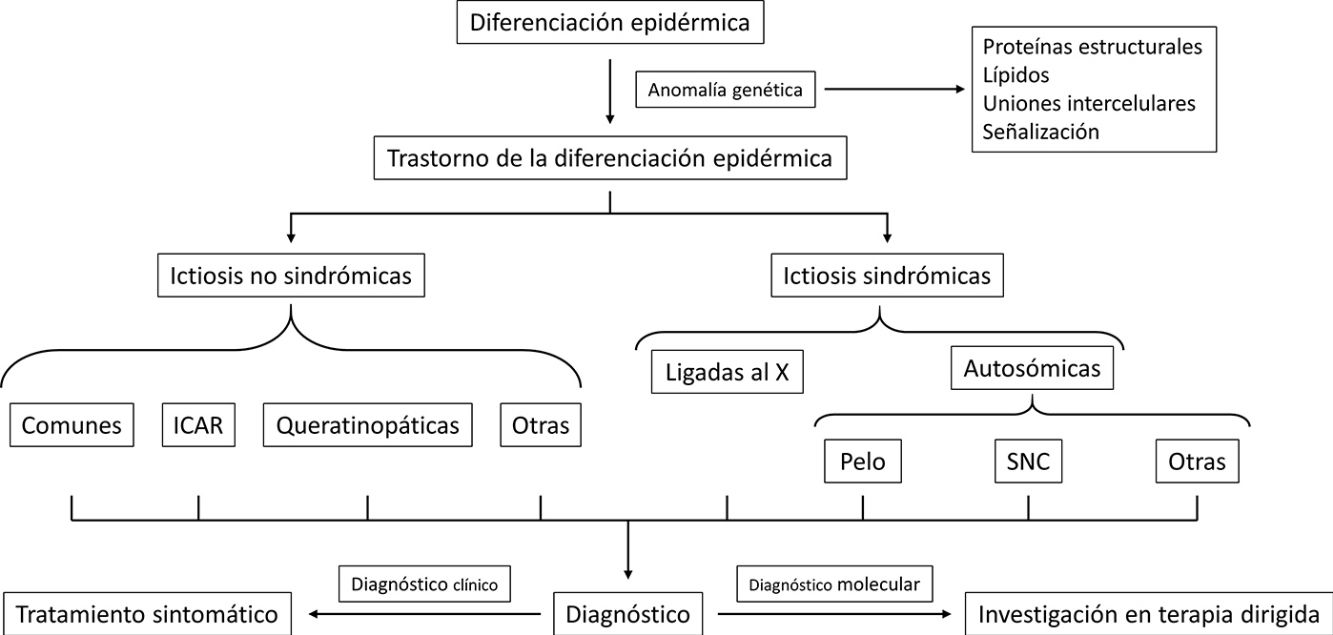


Las ictiosis sindrómicas son un grupo de enfermedades cuyas alteraciones genéticas tienen repercusión tanto en los tejidos epidérmicos como en los no epidérmicos, por lo que los pacientes presentan clínica cutánea y síntomas en varios órganos o aparatos. La mayoría son excepcionales, y en algunas de ellas la descamación ictiosiforme ha sido pobremente caracterizada. Su patrón hereditario es diverso, sus manifestaciones clínicas extracutáneas heterogéneas y la afectación cutánea muy variable, lo cual dificulta una buena subclasificación clínica.
El diagnóstico de las ictiosis reside en una buena anamnesis, una exploración física detallada, y la detección de hallazgos analíticos y/o histológicos asociados. El estudio genético es imprescindible, no solo para alcanzar el diagnóstico de certeza, sino porque conocer el sustrato molecular concreto en cada paciente es el primer paso para encontrar un régimen terapéutico individualizado. El tratamiento pasa por eliminar las escamas y mantener la piel hidratada mediante el uso de terapia tópica con exfoliantes y emolientes. Además, en la actualidad, se están desarrollando terapias de reemplazo dirigidas a sustituir las proteínas y los lípidos específicamente alterados en cada entidad, y se están empezando a evaluar estrategias de terapia génica cuya finalidad última es la curación. En la segunda parte de este trabajo hacemos una actualización clínica y genética de las entidades sindrómicas, así como una actualización del diagnóstico y del tratamiento de las ictiosis.
Syndromic ichthyoses are a group of disorders whose genetic alterations impact both epidermal and non-epidermal tissues. Therefore, patients present symptoms in other organs. Most are extraordinary and, in some, ichthyosiform desquamation has been poorly described. Their patterns of inheritance are diverse; their extracutaneous clinical signs, heterogeneous; and the skin symptoms, highly variable, which hinders a proper clinical classification.
Ichthyosis diagnosis starts with proper anamnesis, detailed physical examination, and detection of associated analytic and/or histologic findings. Genetic testing is indispensable, not only for diagnostic certainty, but also because understanding the molecular substrate for each patient is the first step towards finding an individualized therapeutic regimen. While it will almost invariably involve facilitating desquamation and maintaining skin hydration using topical exfoliants and emollients, recently, replacement therapies aiming at substituting the proteins and lipids specifically altered in each patient are being developed and gene therapy approaches with the ultimate goal of curing the disease are being assessed. In part 2 of this review, we’ll be updating the clinical and genetic findings of syndromic entities, ichthyosis diagnosis and treatment.

Las ictiosis sindrómicas son un grupo de enfermedades cuyas alteraciones genéticas tienen repercusión tanto en los tejidos epidérmicos como en los no epidérmico como consecuencia del trastorno genético, por lo que los pacientes presentan síntomas en otros órganos u aparatos. La mayoría son excepcionales y en algunas de ellas la descamación ictiosiforme ha sido pobremente caracterizada, por lo que solo detallaremos aquí las más relevantes (tabla 1). Se subdividen en función de su herencia y de los síntomas extracutáneos asociados más relevantes1.
Clasificación de las ictiosis sindrómicas
| Ictiosis sindrómicas |
| Ictiosis sindrómicas ligadas al cromosoma X (ORPHA: 281210) |
| Ictiosis recesiva ligada al cromosoma X sindrómica (ORPHA:281090) |
| Síndrome IFAP (ORPHA: 2273) |
| Condrodisplasia punctata tipo 2 (CDPX2, ORPHA: 35173) |
| Síndrome MEND (ORPHA: 401973) |
| Síndrome CHILD (ORPHA: 139) |
| Ictiosis sindrómicas autosómicas con anomalías prominentes del pelo (ORPHA: 281222) |
| Síndrome de Netherton (ORPHA: 634) |
| Síndrome SAM (ORPHA: 369992) |
| Síndrome IHS (ORPHA: 91132) |
| Síndrome ILVASC (ORPHA: 59303) |
| Tricotiodistrofia (ORPHA: 33364) |
| Ictiosis sindrómicas autosómicas con signos neurológicas prominentes (ORPHA: 281238 y ORPHA: 281241) |
| Síndrome de Sjögren - Larsson (ORPHA: 816) |
| Enfermedad de Refsum (ORPHA: 773) |
| Síndrome CEDNIK (ORPHA: 66631) |
| Síndrome MEDNIK (ORPHA: 171851) |
| Queratodermia ictiótica - paraplejia espástica - hipomielinización - facies dismórfica (ORPHA: -) |
| Síndrome de ictiosis congénita - discapacidad intelectual - cuadriplejia espástica (ORPHA: 352333) |
| Síndrome ARC o ARKID (ORPHA: 2697) |
| Enfermedad de Gaucher fetal (ORPHA: 85212) |
| Deficiencia múltiple de sulfatasas (ORPHA: 585) |
| Síndrome de Neu - Laxova (ORPHA: 2671) a arlequín con trombocitopenia (ORPHA: -) |
| Trastornos congénitos de la glicosilación (ORPHA:137) |
| Otras ictiosis sindrómicas (ORPHA: 281244) |
| Síndrome KID (ORPHA: 477) |
| Enfermedad por depósito de lípidos neutros (ORPHA: 98907) |
| Síndrome de ictiosis - prematuridad (ORPHA: 88621) |
| Síndrome de HELIX (ORPHA: 528105) |
| Síndrome de ictiosis - estatura baja - braquidactilia - microesferofaquia (ORPHA: 363992) |
| Queratodermia palmoplantar y perianal / ictiosis similar |
IFAP: ichthyosis follicularis, alopecia, and photophobia (ictiosis folicular, alopecia y fotofobia), MEND: male EBP disorder with neurological defects (desorden EBP masculino con defectos neurológicos), CHILD: congenital hemidysplasia with ichthyosiform erythroderma and limb defects (hemidisplasia congénita con eritrodermia ictiosiforme y defectos en las extremidades), SAM: severe dermatitis, multiple allergies, and metabolic wasting (dermatitis grave, alergias múltiples y desgaste metabólico), ILVASC: ichthyosis, leukocyte vacuoles, alopecia, and sclerosing cholangitis (ictiosis, vacuolas en los leucocitos, alopecia y colangitis esclerosante), CEDNIK: cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma (disgénesis cerebral, neuropatía, ictiosis y queratodermia), MEDNIK: intellectual disability, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma (discapacidad intelectual, enteropatía, sordera, neuropatía periférica, ictiosis y queratodermia), ARC: arthrogryposis, renal dysfunction, and cholestasis (artrogriposis, disfunción renal y colestasis), ARKID: autosomal recessive keratoderma, ichthyosis, and deafness (queratodermia, ictiosis y sordera autosómica recesiva), KID: keratitis, ichthyosis, and deafness (queratitis, ictiosis y sordera), HELIX: hypohidrosis, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia (hipohidrosis, desequilibrio electrolítico, disfunción lacrimal, ictiosis y xerostomía)
En este grupo, todos los genes causales están involucrados en la síntesis de colesterol.
- –
Ictiosis recesiva ligada al cromosoma X sindrómica: los pacientes con este tipo de ictiosis presentan un fenotipo cutáneo similar al de la forma no sindrómica, pero asocian síntomas extracutáneos variables que pueden incluir anosmia, hipogonadismo hipogonadotrófico (síndrome de Kallman) y retraso mental. Se debe a la presencia de microdeleciones heterocigotas amplias que afectan tanto al gen STS2 como a los genes contiguos, cuya ausencia determina el espectro clínico asociado. En sentido estricto, este tipo de ictiosis podría ser considerada no sindrómica, ya que los síntomas extracutáneos no son provocados por la deficiencia de STS, sino por la deleción adicional de genes contiguos que provocan la sintomatología sindrómica.
- –
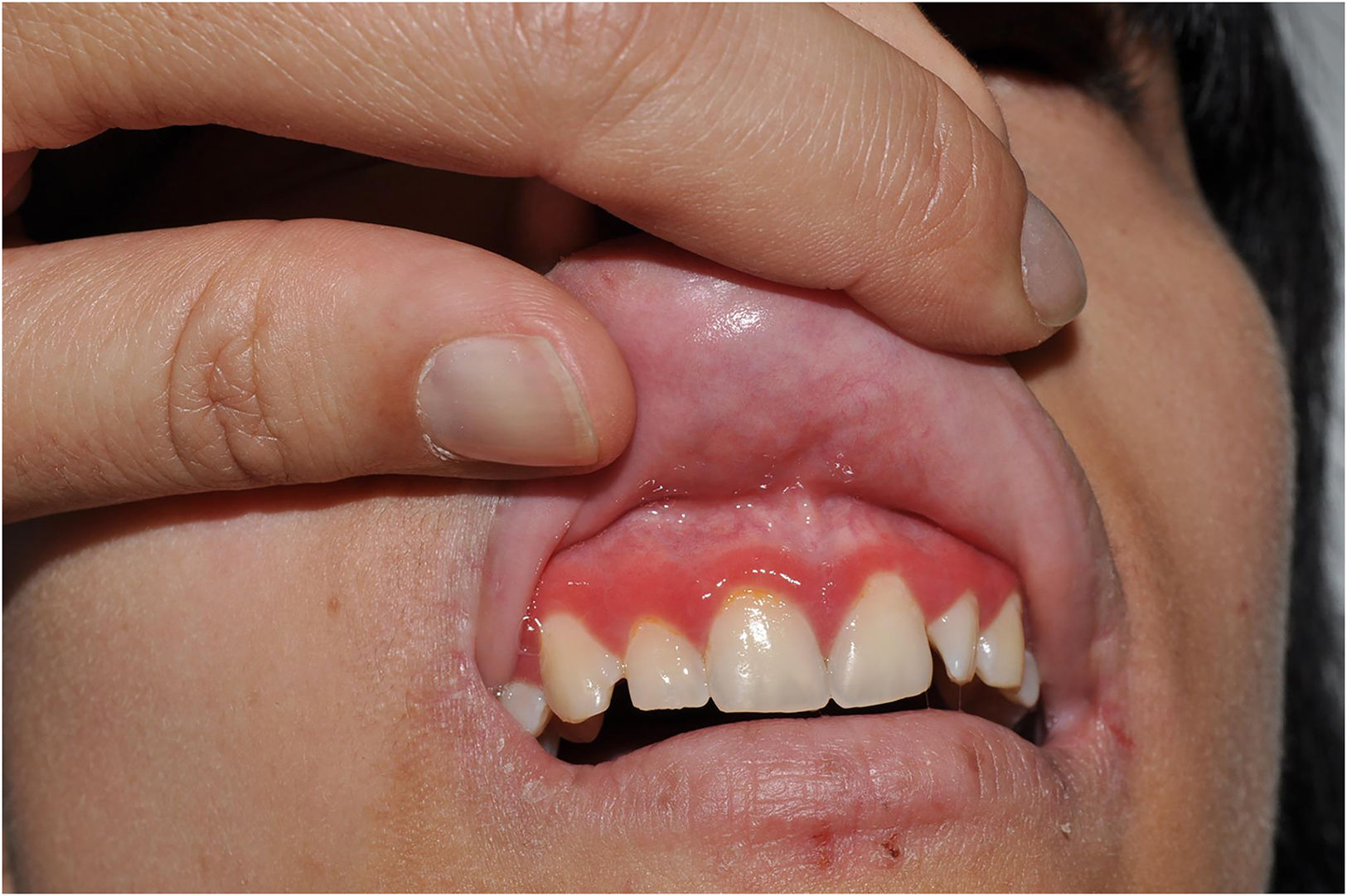
SíndromeIchthyosis follicularis - alopecia – photophobia (IFAP): se caracteriza por afectación cutánea generalizada con eritema, hiperqueratosis folicular, queratodermia palmoplantar, alopecia y fotofobia. Algunos pacientes presentan adicionalmente retraso mental, opacidad corneal, displasia renal, criptorquidia y malformaciones esqueléticas (síndrome BRESHECK). Está causado por variantes patogénicas recesivas en MBTPS23. Es interesante destacar que existe una entidad fenotípicamente muy similar denominada displasia mucoepitelial hereditaria (o IFAP2) originada por variantes patogénicas dominantes en SREBF14, un gen que no se localiza en el cromosoma X sino en el cromosoma 17 (fig. 1). Ambos genes son factores de transcripción que activan la ruta de síntesis de colesterol.
- –
Condrodisplasia punctata tipo 2: esta entidad, letal en los varones, presenta un fenotipo peculiar de ictiosis de distribución blaschkoide debido a inactivación del cromosoma X. Es típica la presencia de cataratas, la asociación de anomalías esqueléticas como talla baja y acortamiento rizomélico de las extremidades, y las imágenes radiológicas puntiformes en las epífisis de los huesos largos Está causada por variantes patogénicas dominantes en EBP5, gen que codifica una enzima de la síntesis de colesterol.
; ictiosis folicular, alopecia y fotofobia) en el paciente con variante patogénica en SREBF1. El eritema gingival es uno de los rasgos distintivos de la enfermedad.")
Este grupo se caracteriza porque los pacientes presentan anomalías características en el pelo, hallazgo clínico que puede facilitar el diagnóstico de la enfermedad. Sin embargo, tanto las manifestaciones clínicas como el sustrato molecular y etiopatogénico es muy diverso.
- –
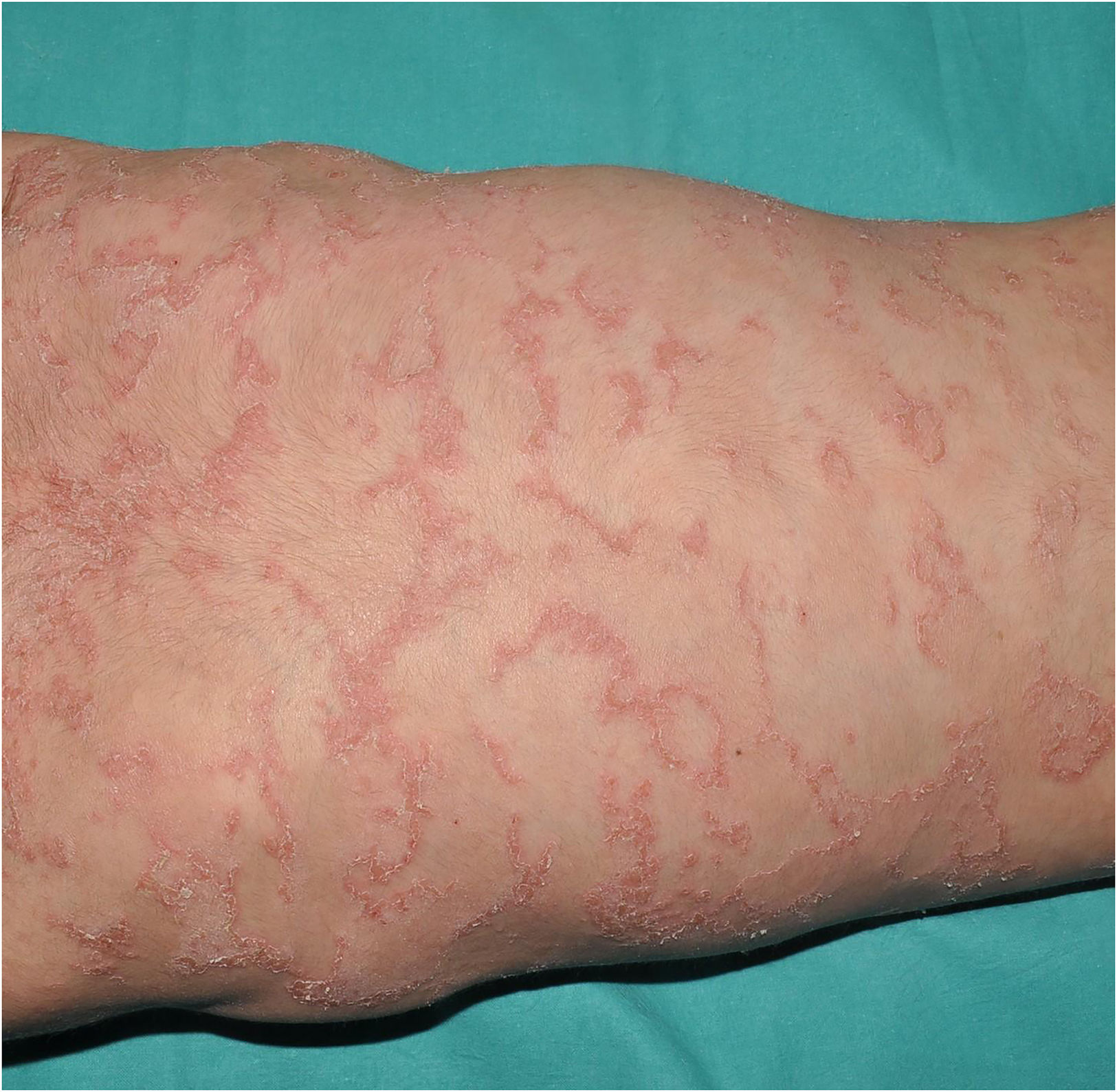
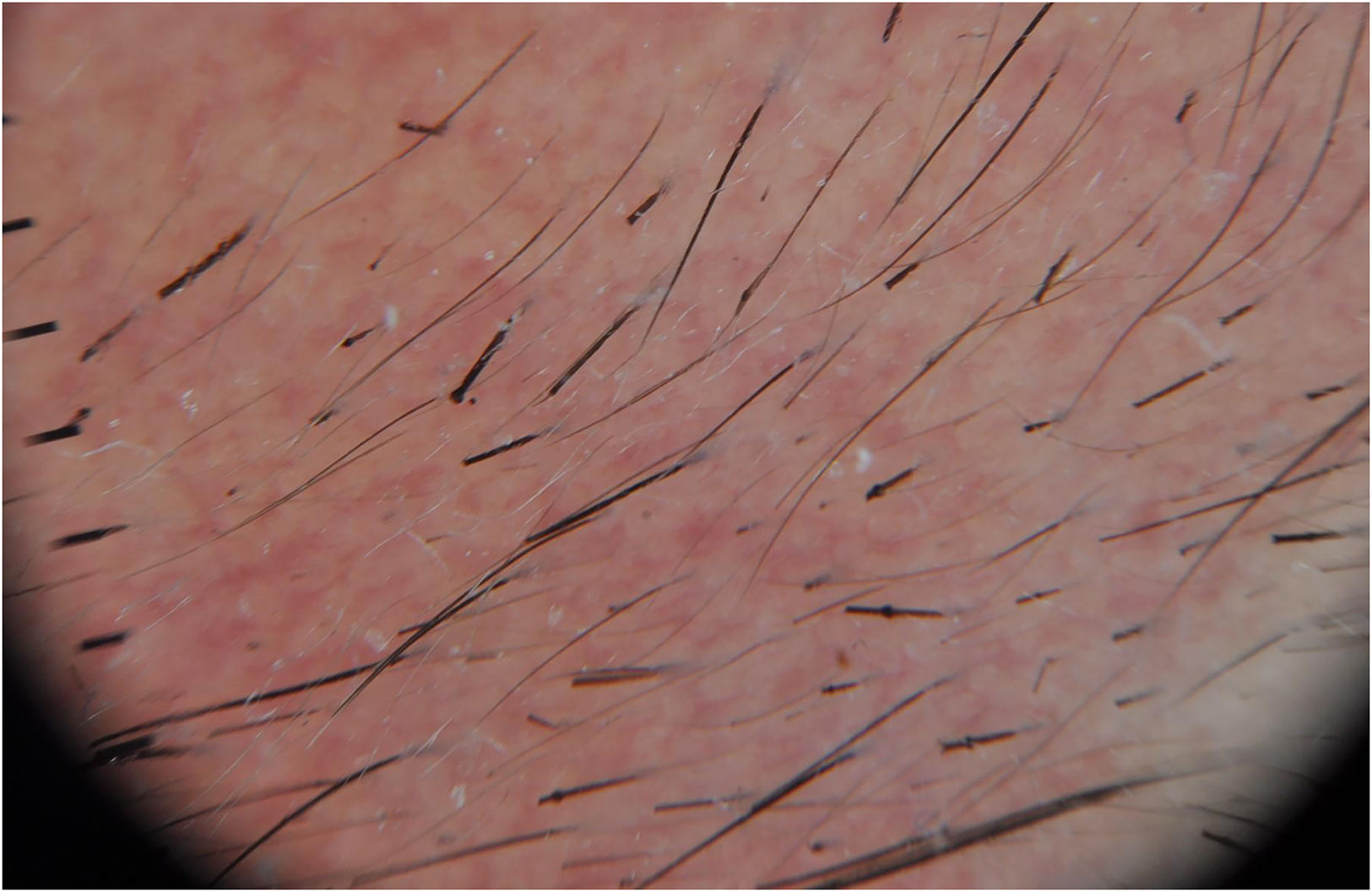
Síndrome de Netherton: es, probablemente, la ictiosis sindrómica más habitual y la que resulta más fácil de diagnosticar cuando se identifican las alteraciones del tallo del pelo. Aunque existen casos descritos de presentación neonatal como bebé colodión, la gran mayoría de los niños nacen eritrodérmicos y con una importante alteración de la barrera cutánea que condiciona la aparición de episodios recurrentes de deshidratación hipernatrémica y sepsis. Es característico el intenso eritema facial de los pacientes, que a menudo presentan un prurito intenso e incoercible. Con el tiempo, los pacientes pueden desarrollar la afectación cutánea característica denominada ictiosis lineal circunfleja, en las que se aprecian lesiones arciformes o polilobuladas con descamación en doble borde (fig. 2). La mayoría de los pacientes presentan tricorrexis invaginata o pelo en caña de bambú, una afectación del tallo del pelo que puede afectar tanto al cabello como a las cejas y las pestañas y que puede adivinarse en la exploración dermatoscópica (fig. 3). En consecuencia, el pelo de los pacientes es corto y desigual, ya que es frágil y se quiebra con facilidad a nivel de las zonas de tricorrexis invaginata. Muchos pacientes sufren también diátesis atópica, presentando brotes eccematiformes en la piel y alergias alimentarias, así como lesiones verruciformes, y predisposición a sufrir infecciones cutáneas por virus del papiloma humano. El síndrome de Netherton está causado por variantes patogénicas recesivas en SPINK56. Se puede diagnosticar por inmunohistoquímica de la piel, ya que los pacientes presentan una ausencia de expresión de LEKTI, la proteína que codifica este gen y que se expresa en el estrato córneo. Su ausencia impide la inactivación de las kalikreinas, enzimas epidérmicas responsables de la degradación de los corneocitos necesaria para la descamación fisiológica.
- –
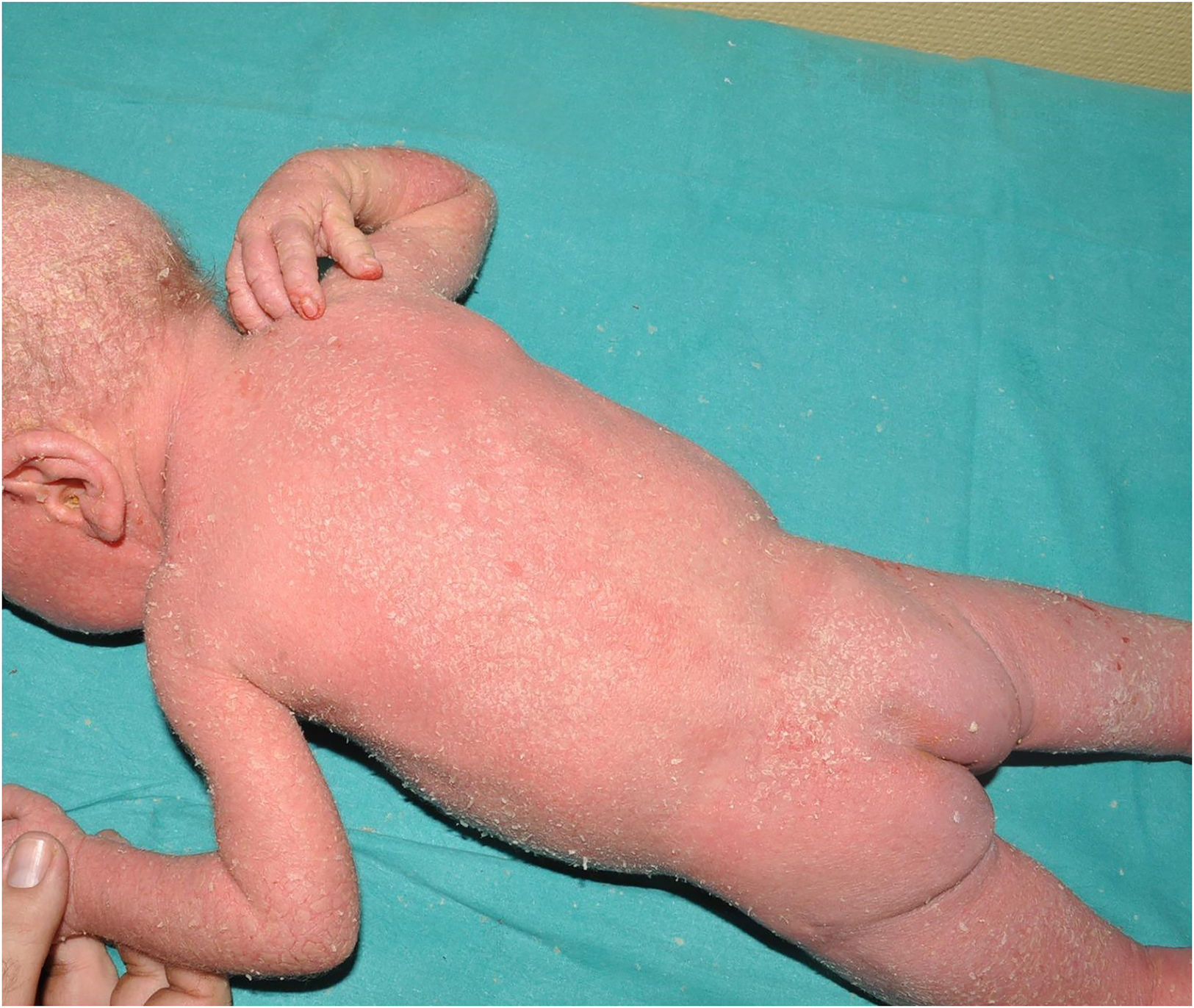
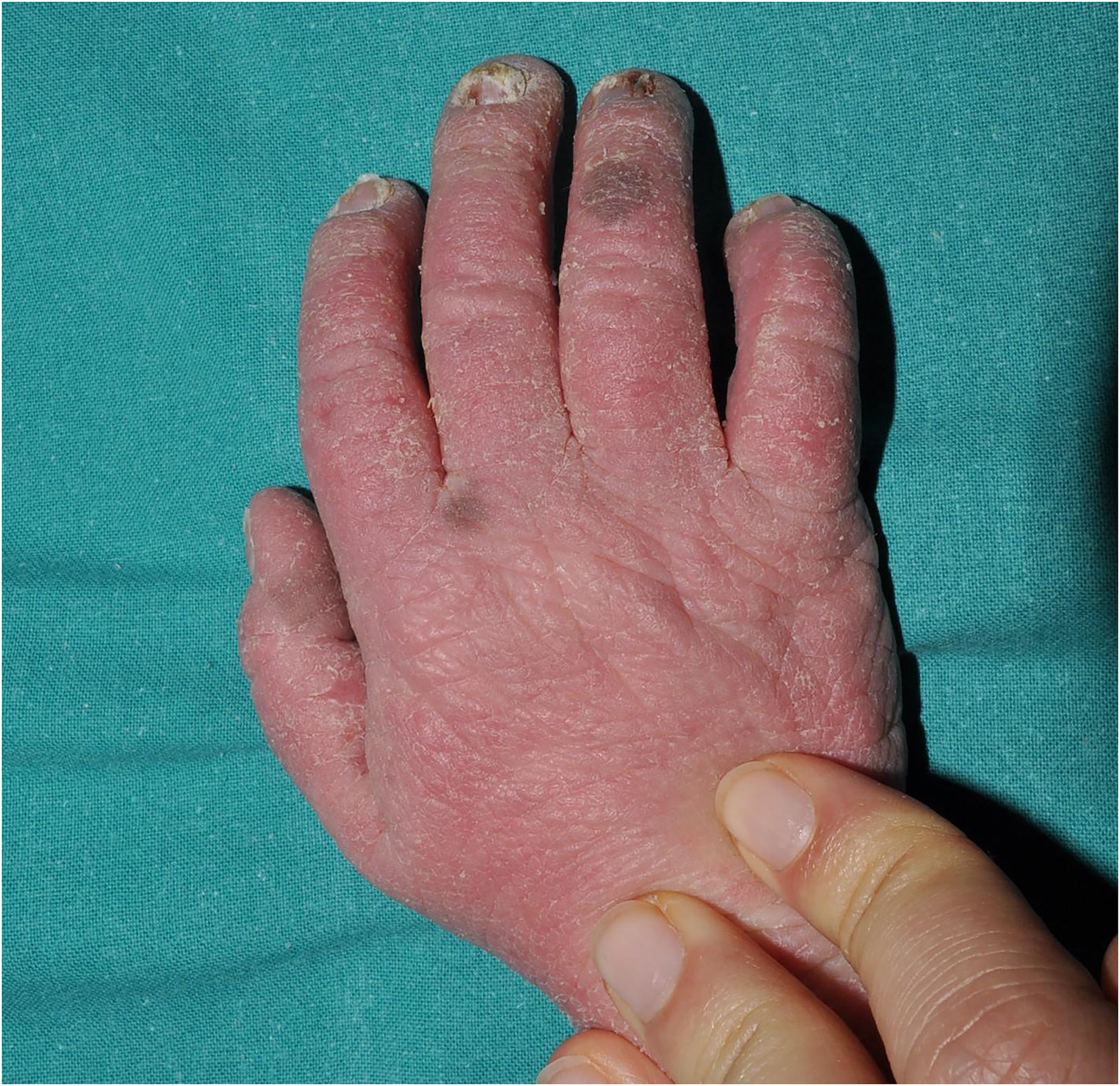
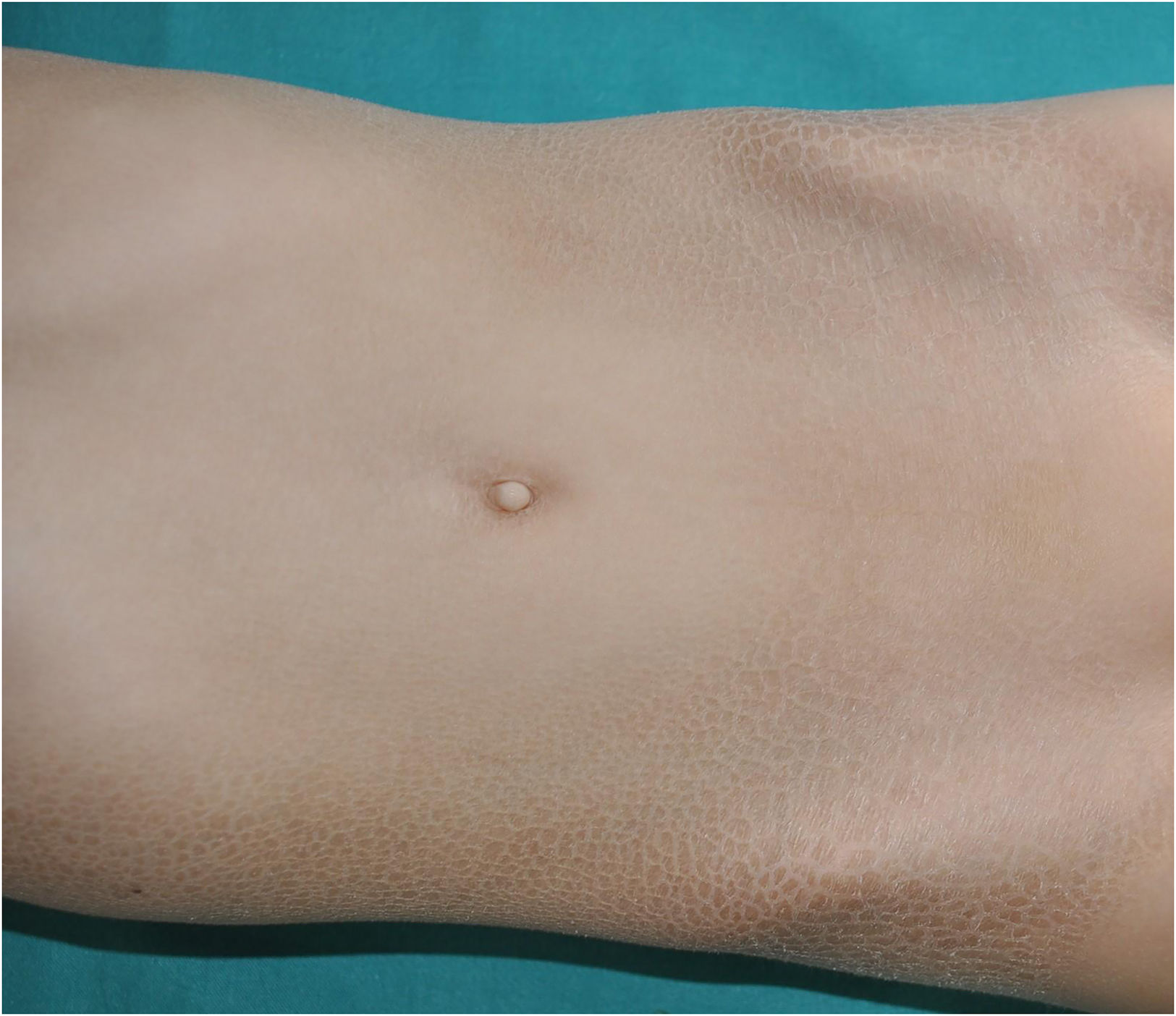
Síndrome Severe dermatitis - multiple allergies - metabolic wasting syndrome (SAM): es un trastorno desmosómico cuyo acrónimo resume sus rasgos clínicos fundamentales: Severe dermatitis (eritrodermia ictiosiforme), Allergies (alergias) y Metabolic wasting, es decir, fallo de medro y retraso estatoponderal grave. Fue descrito tiempo después de la última clasificación consenso, por lo que no está incluida en la misma. Sin embargo, creemos que su origen fisiopatológico y sus manifestaciones clínicas justifican la inclusión del SAM en el grupo de las ictiosis sindrómicas con anomalías del pelo. El cuadro comienza al nacer, y a menudo asocia infecciones sistémicas de repetición que ponen en peligro la vida del niño. Desde el punto de visto cutáneo, los pacientes presentan una eritrodermia descamativa grave con hipotricosis y onicodistrofia (fig. 4). No es infrecuente observar anomalías del esmalte dental y de las lesiones hiperpigmentadas transitorias cuyo análisis histológico sugiere origen postinflamatorio (fig. 5). El síndrome SAM está causado por variantes patogénicas recesivas en DSG17 o dominantes en DSP8. En este último caso, puede haber cardiomiopatía asociada, ya que la DSP se expresa también en el tejido cardiaco9.
; dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia ictiosiforme severa e hipotricosis de cuero cabelludo en un paciente con variante patogénica en DSG1. Obsérvese el picor asociado que provoca el rascado del niño en el hombro derecho.") Figura 4.
Figura 4.Síndrome de Severe dermatitis, multiple allergies, and metabolic wasting (SAM); dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia ictiosiforme severa e hipotricosis de cuero cabelludo en un paciente con variante patogénica en DSG1. Obsérvese el picor asociado que provoca el rascado del niño en el hombro derecho.
(0.34MB).; dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia, onicodistrofia e hiperpigmentación parcheada de naturaleza postinflamatoria en el dorso de los dedos del paciente con variante patogénica en DSP.") Figura 5.
Figura 5.Síndrome de Severe dermatitis, multiple allergies, and metabolic wasting (SAM); dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia, onicodistrofia e hiperpigmentación parcheada de naturaleza postinflamatoria en el dorso de los dedos del paciente con variante patogénica en DSP.
(0.52MB). - –
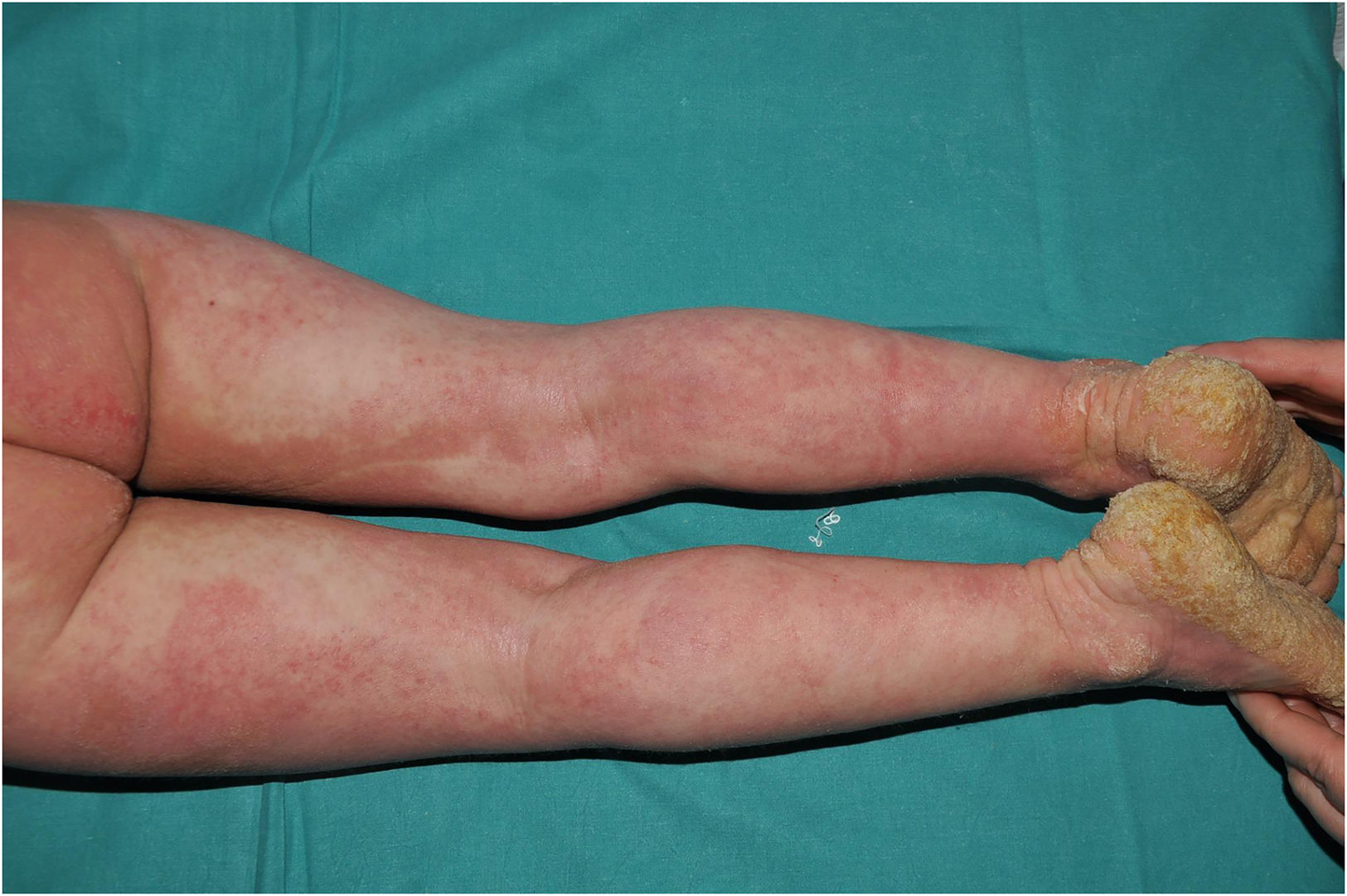
Tricotiodistrofia: se caracteriza por ictiosis junto con pelo y uñas quebradizas en los que se detecta deficiencia de azufre. La descamación ictiosiforme suele ser leve, de tono oscuro y relativamente grande, con afectación de la frente y el tronco (fig. 6). Es característica la observación de bandas alternantes claras y oscuras en el tallo del pelo bajo luz polarizada (bandeado en cola de tigre). Está causada por variantes patogénicas recesivas en AARS110, GTF2E211, MAARS110, MPLKIP12, RNF113A13 o TARS114, que codifican proteínas involucradas en la traducción de proteínas. También existe un subtipo que presenta adicionalmente fotosensibilidad, neuropatía progresiva y envejecimiento acelerado causado por variantes patogénicas recesivas en ERCC215, ERCC316 o GTF2H517, que codifican proteínas encargadas de la reparación del ADN además de la traducción de proteínas. Como en casos anteriores, este grupo presenta una llamativa heterogeneidad molecular que dificulta extraordinariamente la correlación geno y fenotípica.

.")
; dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia ictiosiforme severa e hipotricosis de cuero cabelludo en un paciente con variante patogénica en DSG1. Obsérvese el picor asociado que provoca el rascado del niño en el hombro derecho.")
; dermatitis grave, alergias múltiples y desgaste metabólico. Eritrodermia, onicodistrofia e hiperpigmentación parcheada de naturaleza postinflamatoria en el dorso de los dedos del paciente con variante patogénica en DSP.")

El proceso más frecuente de este grupo es el síndrome de Sjögren-Larsson, que se caracteriza por discapacidad intelectual, parálisis espástica y anomalías oculares (retinopatía cristalina) muy sugerentes del diagnóstico. La afectación cutánea, poco expresiva, consiste en una discreta hiperqueratosis generalizada que en algunas zonas determina lesiones lineales paralelas denominadas en raíl de tren y que en las flexuras puede recordar a la liquenificación. Muchos pacientes presentan picor importante. Está causado por variantes patogénicas recesivas en ALDH3A218, un aldehído graso deshidrogenasa responsable de la síntesis de ácidos grasos utilizados en la síntesis de ceramidas.
Otras ictiosis sindrómicasEste grupo incluye diversas entidades que no responden a las características de los grupos anteriores.
- –
Síndrome keratitis - ichthyosis - deafness (KID): se caracteriza por queratitis, hiperqueratosis espinulosa, queratodermia palmoplantar y sordera neurosensorial. Está causada por variantes patogénicas dominantes en GJB219 o GJB620 o recesivas en AP1B121, que codifican proteínas de las uniones gap encargadas de la comunicación intercelular y el complejo adaptador de las vesículas recubiertas de clatrina respectivamente. Adicionalmente, variantes patogénicas poscigóticas en GJB2 causan lesiones queratósicas siguiendo una distribución blasckoide, que si afectan a las gónadas pueden transmitirse a la descendencia como síndrome KID22. Estas formas mosaico tienen un menor impacto sistémico porque respetan la visión y la audición (fig. 7).
; queratitis, ictiosis y sordera mosaico. Afectación en las líneas de Blaschko de las extremidades inferiores en el paciente con variante patogénica en GJB2 de aparición poscigótica. Obsérvese la importante queratodermia plantar asociada.") Figura 7.
Figura 7.Síndrome keratitis, ichthyosis, and deafness (KID); queratitis, ictiosis y sordera mosaico. Afectación en las líneas de Blaschko de las extremidades inferiores en el paciente con variante patogénica en GJB2 de aparición poscigótica. Obsérvese la importante queratodermia plantar asociada.
(0.22MB). - –
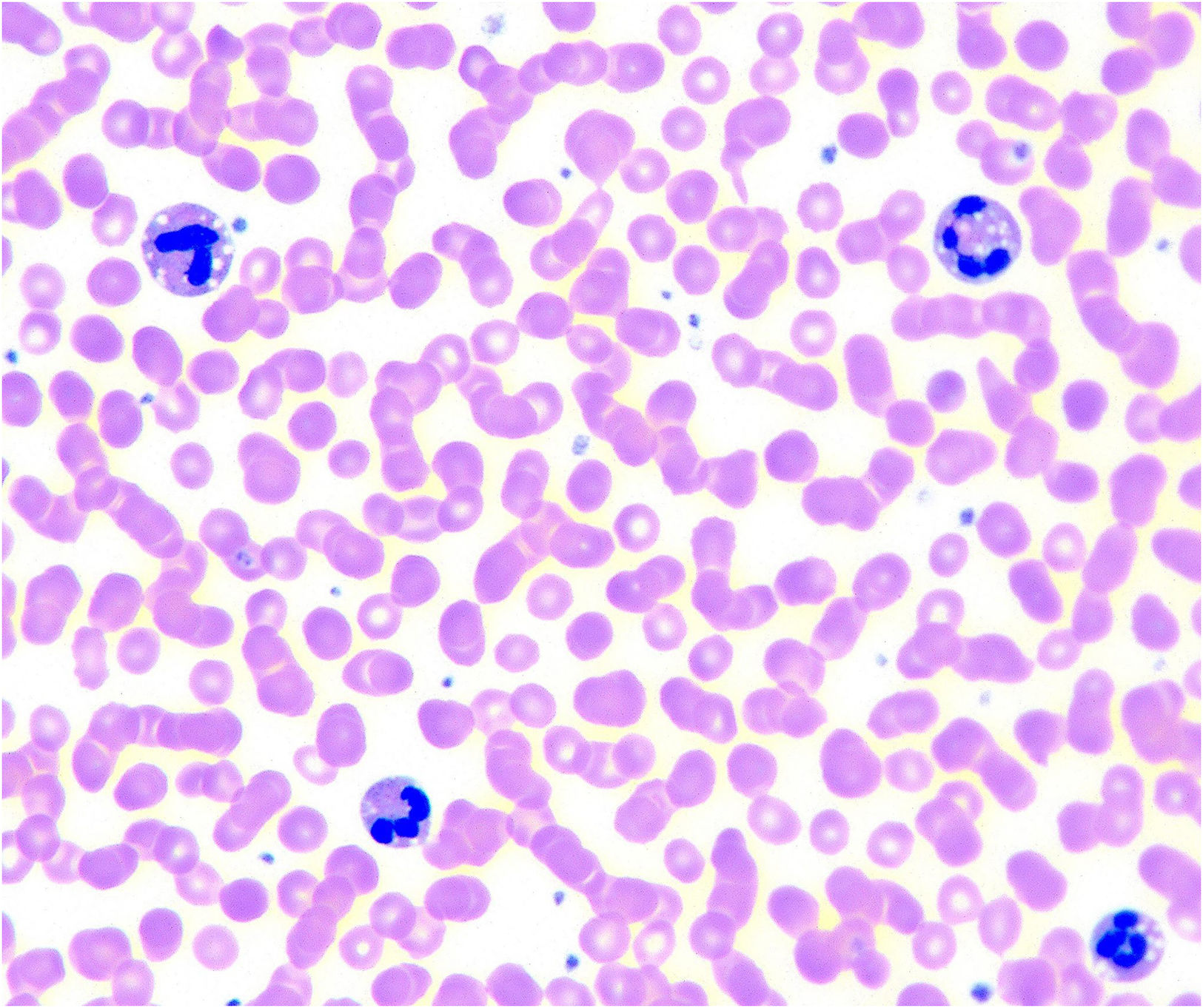
Enfermedad por depósito de lípidos neutros: también conocida como enfermedad de Chanarin-Dorfman, este cuadro se caracteriza por ictiosis con fenotipo de eritrodermia ictiosiforme congénita leve, hepatoesplenomegalia, miopatía, hipoacusia y cataratas23. Histológicamente, se observa acumulación de vacuolas lipídicas en la mayoría de tejidos (fig. 8). Está causada por variantes patogénicas recesivas en ABHD524, que codifica una enzima que sintetiza ácido linoleico para la síntesis de ceramidas y cuya anomalía determina el depósito de ácidos grasos neutros intracelulares.
; queratitis, ictiosis y sordera mosaico. Afectación en las líneas de Blaschko de las extremidades inferiores en el paciente con variante patogénica en GJB2 de aparición poscigótica. Obsérvese la importante queratodermia plantar asociada.")
.")
Las características principales de las ictiosis sindrómicas más infrecuentes se detallan en la tabla 2.
Formas infrecuentes de las ictiosis sindrómicas
| Enfermedad | Genes causales | Características principales |
|---|---|---|
| Síndromes con ictiosis ligada al X (ORPHA: 281210) | ||
| Trastorno EBP masculino con defectos neurológicos (MEND, ORPHA: 401973) | EBP1 (XR, MIM: 300960) | Caracterizado por ictiosis, síntomas neurológicos (retraso en el desarrollo y convulsiones) y dismorfia craniofacial, con posible afectación de otros órganos1. |
| Hemidisplasia congénita con nevo ictiosiforme y defectos en las extremidades (CHILD, ORPHA: 139) | NSDHL2 (XD, MIM: 308050) | Caracterizada por un nevo principalmente ipsilateral (afectando estrictamente a la mitad del cuerpo a lo largo del plano sagital) con hipoplasia de las estructuras esqueléticas (baja estatura o ausencia de extremidades) y, en algunos pacientes, del cerebro y los órganos (pulmones, corazón y riñones)2. |
| Síndromes ictióticos autosómicos (con) | ||
| Anomalías prominentes del pelo (ORPHA: 281222) | ||
| Síndrome de ictiosis - hipotricosis (IHS, ORPHA: 91132) | ST143 (AR, MIM: 602400) | También conocido como síndrome de ictiosis - atrofodermia - hipotricosis - hipohidrosis. Se caracteriza por ictiosis congénita difusa, indentaciones foliculares sin pelo (atrofodermia folicular), hipotricosis e hipohidrosis3. |
| Síndrome de ictiosis - leucocitos vacuolados - alopecia - colangitis esclerosante (ILVASC, ORPHA: 59303) | CLDN14 (AR, MIM: 607626) | También conocido como síndrome de ictiosis neonatal - colangitis esclerosantes. Se asocia a ictiosis, hipotricosis del cuero cabelludo, alopecia cicatrizal, anomalías dentales e inflamación de los conductos biliares4. |
| Signos neurológicos prominentes (ORPHA: 281238 and ORPHA: 281241) | ||
| Enfermedad de Refsum (ORPHA: 773) | PEX75 (AR, MIM: 308100), PHYH6,7 (AR, MIM: 266500) | También conocida como neuropatía motor y sensorial hereditaria tipo 4, heredopatía atáctica polineuritiforme o deficiencia de fitanico - CoA hidroxilasa. Se caracteriza por pérdida progresiva de función retinal (retinitis pigmentosa), neuropatía periférica, falta de olfato (anosmia), falta de coordinación motora (ataxia cerebelosa) e ictiosis. La elevación (>200μmol/l) de los niveles de ácido fitánico en suero es patognómica5. |
| Síndrome de disgenesia cerebral - neuropatía - ictiosis - queratodermia palmoplantar (CEDNIK, ORPHA: 66631) | SNAP298 (AR, MIM: 609528) | Suele causar muerte prematura por neumonía por aspiración8. |
| Síndrome de discapacidad intelectual - enteropatía - sordera - neuropatía periférica - ictiosis - queratodermia (MEDNIK, ORPHA: 171851) | AP1S19 (AR, MIM: 609313), AP1B110 (AR, MIM: 242150) | Causado por variantes patogénicas recesivas en AP1S1.Un síndrome fenotípicamente similar, llamado síndrome autosómico recesivo de queratitis - ictiosis - sordera (KIDAR, ORPHA: -), está causado por variantes patogénicas recesivas en AP1B1. |
| Queratodermia ictiótica - paraplejía espástica - hipomielinización - facies dismórfica (ORPHA: -) | ELOVL111 (AD, MIM: 618527) | Caracterizada por queratodermia ictiótica, paraplejía espástica, hipomielinización y facies dismórfica. |
| Síndrome de ictiosis congénita - discapacidad intelectual - cuadriplejía espástica (ORPHA: 352333) | ELOVL412 (AR, MIM: 614457) | También conocido como neuroictiosis asociada al gen ELOVL4, esta enfermedad también presenta convulsiones |
| Síndrome de artrogriposis - disfunción renal - colestasis (ARC, ORPHA: 2697) | VIPAS3913 (AR, MIM: 613404), VPS33B14 (AR, MIM: 208085) | Caracterizado por artrogriposis neurogénica, disfunción de los túbulos renales, defectos en la producción de bilis (colestasis) ictiosis y muerte en el primer año de vida14.Esta enfermedad es alélica con queratodermia - ictiosis - sordera - autosómica recesiva (ARKID, ORPHA: -), también causada por mutaciones patogénica recesivas en VPS33B15. |
| Enfermedad de Gaucher fetal (FGD, ORPHA: 85212) | GBA116 (AR, MIM: 608013) | También llamada enfermedad de Gaucher tipo II o perinatal letal. Se caracteriza por movilidad fetal reducida, contractura de las articulaciones (artrogriposis), dismorfia facial, con trombocitopenia ocasional, ictiosis y muerte en útero o poco después del nacimiento16. Estos neonatos y niños experimentan deterioro neurológico progresivo. |
| Deficiencia múltiple de sulfatasas (MSD, ORPHA: 585) | SUMF117 (AR, MIM: 272200) | También conocida como sulfatidosis juvenil tipo Austin. Se caracteriza por ictiosis similar a IRLX, retraso en el desarrollo y las anomalías neurológicas y esqueléticas de las enfermedades de almacenamiento y muerte prematura debida a complicaciones respiratorias18. |
| Síndrome de Neu-Laxova (NLS, ORPHA: 2671) | PHGDH19 (AR, MIM: 256520), PSAT120 (AR, MIM: 616038), PSPH21 (AR, MIM: -) | Caracterizado por membrana colodión, malformaciones severas, microcefalia y retraso en el crecimiento intrauterino que lleva a muerte in utero o poco después del nacimiento21. |
| Trastornos de la glicosilación | ||
| Deficiencia de UDP - glucosa ceramida glicosiltransferasa (ORPHA: -) | UGCG22 (AR, MIM: -) | Se manifiestan como bebé colodión con contracturas congénitas de las articulaciones22. Esta condición descrita recientemente ha sido mayoritariamente letal en los primeros meses de vida, pero se espera que tuviera efectos neurológicos graves22. |
| Trastorno congénito de la glicosilación tipo 1F (CDG-1F, ORPHA: 79323) | MPDU123 (AR, MIM: 609180) | Caracterizado por tono muscular excesivo (hipertonía), retraso psicomotor e ictiosis23. |
| Trastorno congénito de la glicosilación tipo 1M (CDG-1M, ORPHA: 91131) | DOLK24 (AR, MIM: 610768) | También conocido como deficiencia de dolicol quinasa o hipotonía con ictiosis debido a deficiencia de fosfatasa de dolicol. Se caracteriza por reducción del tono muscular (hipotonía), inflamación, cardiomiopatía frecuente e ictiosis24. |
| Trastorno congénito de la glicosilación tipo 1Q (CDG-1Q, ORPHA: 324737) | SRD5A325 (AR, MIM: 612379) | Presenta colobomas oculares, malformaciones cerebrales con discapacidad intelectual, hiperplasia de la glándula pituitaria e ictiosis25. |
| Síndrome de coloboma - enfermedad cardíaca congénita - dermatosis ictiosiforme - discapacidad intelectual - anomalías de las orejas (CHIME, ORPHA: 3474) | PIGL26 (AR, MIM: 280000) | Presenta coloboma, enfermedad cardíaca congénita, dermatosis ictiosiforme, discapacidad intelectual y anomalías de las orejas26. |
| Otros signos asociados (ORPHA: 281244) | ||
| Síndrome de ictiosis - prematuridad (IPS, ORPHA: 88621) | SLC27A427 (AR, MIM: 608649) | Caracterizado por nacimiento prematuro, asfixia neonatal y placas ictióticas adoquinadas con extensa descamación que recuerda a la vernix27 y suele mejorar drásticamente durante el periodo neonatal hasta dar lugar a piel casi normal. Los niños nacen con prematuridad cubiertos con una gruesa película parecida al vérmex especialmente expresiva en el macizo facial |
| Síndrome de hipohidrosis - desequilibrio electrolítico - disfunción lacrimal - ictiosis - xerostomía (HELIX, ORPHA: 528105) | CLDN1028 (AR, MIM: 617671) | Caracterizado por hipohidrosis, pérdida renal de iones Na+ y Cl- causando desequilibrio electrolítico, ojo y boca secos (xeroftalmia y xerostomía) e ictiosis28. |
| Síndrome de ictiosis - estatura baja - braquidactilia - microesferofaquia (ORPHA: 363992) | CERS329 + ADAMTS1729 (HD, MIM: -) | También conocido como síndrome de microdeleción del 15q26.3. Se caracteriza por estatura baja, dedos cortos (braquidactilia), anomalías del cristalino (microesferofaquia) y miopía, características del síndrome de Weill - Marchesani (asociado a ADAMTS17), así como ictiosis con fenotipo EIC (asociado a CERS3)29. |
| Queratodermia palmoplantar y perianal / ictiosis similar a arlequín con trombocitopenia (ORPHA: -) | KDSR30 (MIM: -) | Los pacientes presentan trombocitopenia con, o bien hiperqueratosis en palmas, suelas y piel anogenital, o bien síntomas cutáneos similares a la ictiosis arlequín |
AD: autosómico dominante, AR: autosómico recesivo, HD: deleción homocigota, XD: dominante ligado al X, XR: recesivo ligado al X
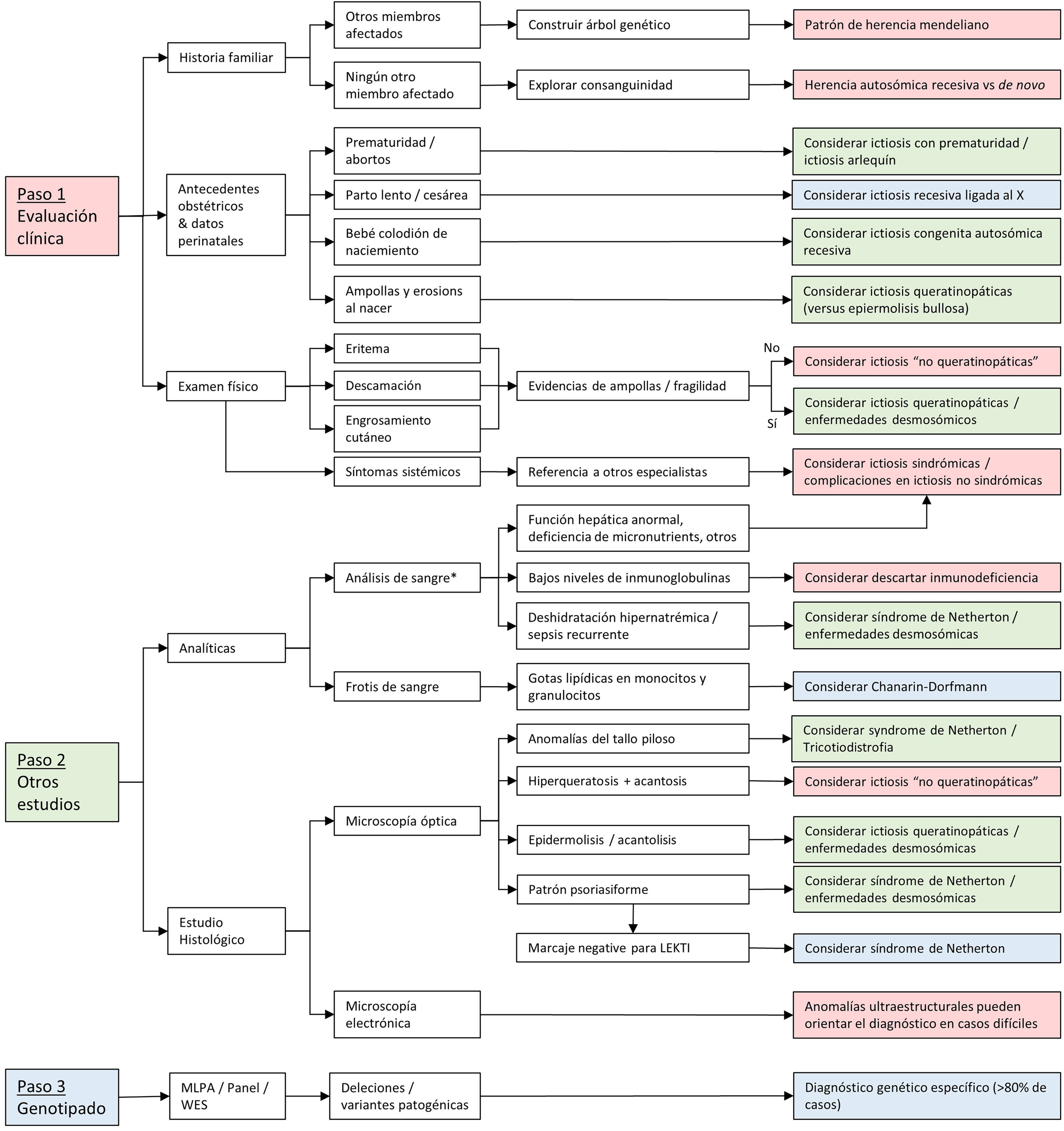
La alteración generalizada de la barrera cutánea en los pacientes con trastornos de la diferenciación epidérmica (TDE) está presente desde el nacimiento o los primeros meses de vida, lo que facilita el diagnóstico clínico. Sin embargo, la escasa correlación entre el fenotipo y el genotipo dificulta un diagnóstico más preciso en muchos casos. En la figura 9 se propone un algoritmo diagnóstico en el que una adecuada anamnesis es determinante.

Las analíticas de sangre, incluyendo recuento de glóbulos rojos, funciones hepática y renal, niveles de electrolitos en sangre, niveles de inmunoglobulinas en suero y frotis de sangre, pueden ayudar a excluir formas sindrómicas de ictiosis con sus anomalías asociadas25. Por ejemplo, el síndrome de Netherton y los trastornos desmosómicos llevan asociado un riesgo de deshidratación hipernatrémica en bebés7,26. Además, los niveles de inmunoglobulinas en suero pueden ser útiles en el diagnóstico diferencial con inmunodeficiencias hereditarias, que también pueden presentar eritema y descamación. Se debe considerar referir el paciente a otros especialistas dependiendo de los hallazgos clínicos25. El síndrome Chanarin-Dorfman presenta gotas lipídicas en granulocitos y monocitos en frotis de sangre (la denominada anomalía de Jordan)27 (fig. 8).
La realización de biopsias para histología de rutina, inmunohistoquímica o, más raramente, microscopía electrónica pueden ayudar en el diagnóstico diferencial28. La ausencia de expresión inmunohistoquímica de LEKTI (codificada por SPINK5) puede confirmar un diagnóstico de síndrome de Netherton29 y es de especial utilidad si no se dispone de análisis genético28. El examen del tallo del pelo al microscopio óptico es una técnica accesible y no invasiva que aporta información útil en las ictiosis asociadas a anomalías capilares, como la tricorrexis invaginata en el síndrome de Netherton o la apariencia en «cola de tigre» bajo luz polarizada en la tricotiodistrofia30. A pesar de que el diagnóstico clínico de las ictiosis puede realizarse fácilmente, la correlación fenotipo-genotipo es a menudo difícil de establecer. El análisis genético por secuenciación de última generación es de uso generalizado en países desarrollados para confirmar el diagnóstico, pero no encuentra anomalías genéticas entre un 15 y un 20% de los pacientes con fenotipo de TDE31-35. Esto puede deberse a variantes patogénicas no detectables o no descritas, siendo estas últimas las que permiten ampliar el grupo de las ictiosis cuando se correlacionan con hallazgos clínicos distintivos.
Consejo genéticoLos TDE siguen patrones de herencia mendeliana monogénica, lo que permite el consejo genético, en el que se facilita información a las familias afectadas (pacientes y familiares) sobre los mecanismos moleculares y la probabilidad de transmisión a posibles descendientes. Sin embargo, la percepción del riesgo de tener un hijo afectado varía mucho dependiendo de cada enfermedad y un embarazo puede no ser considerado de riesgo por parejas que ya tienen un hijo con afectación leve. Por el contrario, los pacientes con formas graves o afectación importante de la calidad de vida pueden pedir consejo genético para evitar riesgos en futuros embarazos36. Particularidades regionales, culturales y socioeconómicas también pueden influir en el acceso al consejo genético.
El diagnóstico prenatal requiere de la obtención de tejido embriónico. Bajos niveles de estriol no conjugado y variación en número de copias en suero materno (que detecta deleciones en los cromosomas sexuales maternos) están estrechamente relacionados con un aumento del riesgo de IRLX en fetos varones y puede utilizarse, junto con otras técnicas moleculares, para el diagnóstico prenatal37,38. En cualquier caso, el método de elección para diagnóstico prenatal es el análisis genético de variantes patogénicas conocidas en la familia. El diagnóstico preimplantacional puede ser utilizado por parejas en riesgo para elegir embriones no afectados antes de la fecundación in vitro, aunque muchos países no lo consideran apropiado para ictiosis39.
TratamientoLos TDE son enfermedades genéticas que todavía no tienen tratamiento curativo. Actualmente la terapia está dirigida a aliviar los síntomas con el fin de mejorar la calidad de vida de los pacientes. Esto requiere tratamientos que, por tanto, no solo tienen que ser efectivos sino también seguros y bien tolerados. Sin embargo, la realidad es que hay pocos estudios sobre los efectos a largo plazo de las terapias y su eficacia varía mucho entre los pacientes, incluso entre los que padecen el mismo tipo de TDE. Por tanto, el tratamiento elegido y el régimen de administración dependen de recomendaciones de expertos recogidas en guías de atención, del intercambio de información con asociaciones de pacientes, de la accesibilidad al tratamiento y de la experiencia personal de cada paciente26,40,41.
Tratamiento tópicoLa base de toda terapia de los TDE son los tratamientos tópicos, cuyo objetivo es disminuir el grosor de la epidermis y facilitar su descamación, así como reducir el prurito, la tirantez y la formación de fisuras, mejorando la apariencia de la piel. Para ello pueden actuar a distintos niveles, pero el objetivo siempre es normalizar la función de la barrera epidérmica42.
Baños y exfoliaciónLos baños tienen como objetivo ablandar las escamas para facilitar su eliminación mediante la exfoliación mecánica. Los baños han de ser prolongados y se pueden suplementar con sales, aceites o bicarbonato para aumentar la hidratación y promover la exfoliación. El hipoclorito sódico diluido puede ayudar a controlar las infecciones cutáneas y disminuir el olor que puede provocar el sobrecrecimiento microbiano en los pacientes con hiperqueratosis pronunciada. La exfoliación se puede llevar a cabo con la mano, esponjas, limas, piedra pómez o incluso con tijeras en el caso de las escamas más grandes con desprendimiento parcial. Este proceso suele requerir entre 30 y 60 min al día, lo cual es un factor que incide significativamente en la calidad de vida de los pacientes, ya que deben priorizar esto sobre otras actividades como los juegos infantiles, el estudio, el trabajo y el ocio. En los TDE con afectación del cuero cabelludo es importante eliminar las escamas y costras para evitar la alopecia cicatricial, que muchas veces es inevitable a pesar de estos cuidados.
EmolientesLos emolientes tienen como objetivo mantener un correcto equilibrio hídrico, hidratando la epidermis y sellando la barrera para evitar la pérdida de agua. Su formulación influye en el grado de hidratación, lubricación y oclusión que proporcionan. Sin embargo, el efecto ocluyente puede interferir con la sudoración, agravando la intolerancia al calor que sufren muchos pacientes. La vaselina y la parafina son emolientes baratos y eficaces, pero cosméticamente incómodos, lo que puede hacer preferibles otras formulaciones basadas en urea, propilenglicol o dexpantenol. En general, suele ser necesario aplicarlos por lo menos 2 veces al día40.
Queratolíticos tópicosLos queratolíticos facilitan la degradación proteolítica del citoesqueleto y las uniones intercelulares, lo cual reduce la hiperqueratosis y facilita la descamación. Estos fármacos incluyen urea, ácido salicílico y ácido láctico. Los efectos adversos suelen ser leves, e incluyen picor, ardor e irritación; no obstante, los ácidos pueden absorberse y tener efectos secundarios sistémicos43. Esto es especialmente grave en el caso del ácido salicílico, que puede causar salicilismo con náuseas, vómitos, taquipnea, irritabilidad, coma e incluso muerte. Por tanto, es especialmente importante limitar su aplicación, particularmente en niños, pacientes con función renal o hepática alterada y en casos con un elevado porcentaje de la superficie corporal afectada44.
RetinoidesLos retinoides disminuyen la proliferación y diferenciación de los queratinocitos, controlando el número de células que pueden formar el estrato córneo y reduciendo la hiperqueratosis y la inflamación. Están disponibles solo en algunos países en los que están comercializados para el acné, por lo que se emplean fuera de indicación en ficha técnica. El tazaroteno45 y el adapaleno46 han demostrado eficacia con baja incidencia de efectos adversos47.
En el caso de los pacientes con síntomas más graves, el tratamiento tópico aislado puede requerir mucho tiempo y dar resultados subóptimos, lo que lleva al uso de retinoides orales. Los más utilizados son la acitretina, alitretinoína, y la isotretinoína. Son de especial utilidad en enfermedades con escamas gruesas como las de la ictiosis laminar, pero son poco eficaces en formas puramente eritrodérmicas y mal tolerados por los pacientes con fragilidad cutánea como las ictiosis epidermolíticas. Los efectos secundarios de los retinoides sistémicos suelen ser dosis dependientes e incluyen queilitis, sequedad nasal, xerosis, pérdida de pelo, irritación de la conjuntiva y anomalías lipídicas y hepáticas. Su uso crónico puede causar hiperostosis esquelética con espolones y calcificaciones de la columna vertebral y los tendones. Adicionalmente, tienen un prolongado potencial teratogénico con periodos de eliminación que varían entre un mes (isotretinoína y alitretinoína) y 3 años (acitretina). Por tanto, es necesario realizar un seguimiento a los pacientes que siguen tratamientos con retinoides orales que incluya enzimas hepáticas, perfil lipídico, radiografías y test de embarazo. Estas consideraciones junto con la edad condicionan el agente utilizado, así como las dosis iniciales y de mantenimiento a utilizar48-50.
Terapias dirigidasRecientemente están empezando a aparecer terapias dirigidas de manera más específica contra las alteraciones moleculares de los TDE.
Una de estas dianas es la disregulación inmune, ya que diversos TDE muestran polarización hacia un fenotipo Th17/IL-23. Consecuentemente, se han empezado a utilizar anticuerpos monoclonales dirigidos contra diferentes proteínas de esta vía como IL-17A (secukinumab), IL-12 e IL-23 (ustekinumab) e IL4R/13R (dupilumab). Estos anticuerpos han demostrado ser útiles en algunos pacientes con trastornos desmosómicos51,52 o síndrome de Netherton, pero su eficacia irregular y/o transitoria no permite generalizar su uso53.
Las terapias de reemplazo van dirigidas contra las propias proteínas y lípidos alterados en cada TDE con el objetivo de suministrar las moléculas alteradas en cada enfermedad. La terapia de reemplazo de lípidos ha mostrado especial utilidad en el síndrome CHILD en el que el tratamiento con colesterol y estatinas mejora marcadamente el fenotipo de la mayoría de pacientes54,55. Paralelamente, se está evaluando terapia de reemplazo proteico para suministrar transglutaminasa 1 en los pacientes con ictiosis laminar56.
La terapia génica tiene como objetivo suplementar una copia sana del gen afectado (terapia génica farmacológica) o reparar la mutación en el ADN genómico del paciente (terapia génica curativa). Hasta la fecha solo se han evaluado estrategias de terapia génica farmacológica en ensayos clínicos dirigidos contra TGM1 en ictiosis laminar57 y SPINK5 en síndrome de Netherton57. La terapia génica curativa está en los primeros estadios de desarrollo, debido a la dificultad para modificar el ADN genómico, pero hay un gran interés ya que sería el único tratamiento capaz de curar una enfermedad genética58.
Aspectos especiales del tratamientoMuchos TDE presentan síntomas asociados a los defectos de la barrera cutánea que también tienen que ser controlados. El prurito es muy común y disminuye considerablemente la calidad de vida59. El tratamiento con anticuerpos monoclonales contra las rutas inflamatorias (dupilumab60, ustekinumab61 y secukinumab52) parecen eficaces, pero los resultados son inconstantes y a menudo transitorios. La hipohidrosis aumenta el riesgo de sufrir golpes de calor, lo que hace necesario limitar el ejercicio físico y permanecer en entornos frescos. En el cuidado ocular, el principal objetivo es mantener la lubricación mediante el uso profiláctico de lágrimas artificiales62. La descamación en el canal auditivo puede causar hipoacusia de conducción, aumenta el riesgo de infecciones y puede causar daño irreversible en el tímpano, lo que hace necesario el seguimiento otorrinolaringológico regular63. En las formas más graves, es preciso un seguimiento regular de parámetros metabólicos y una suplementación nutricional adecuada64. El defecto de la barrera epidérmica facilita la aparición de infecciones bacterianas y fúngicas, que pueden pasar desapercibidas debido al fenotipo cutáneo de los TDE.
Los síntomas extracutáneos de los TDE ponen de manifiesto la necesidad crítica de equipos multidisciplinares para proporcionar un cuidado adecuado. Oftalmólogos, otorrinolaringólogos y nutricionistas son especialmente importantes. Los pacientes con manifestaciones extracutáneas necesitan especialistas de acuerdo a los órganos afectados, como neurólogos, gastroenterólogos o traumatólogos. Así, numerosos pacientes con IRLX presentan déficit de atención e hiperactividad y necesitan atención neuropsicológica65. Finalmente, algunos estudios han mostrado que la fisioterapia puede disminuir la gravedad de los síntomas y mejorar la calidad de vida66.
Impacto en la calidad de vidaLos TDE pueden tener un impacto profundo en la calidad de vida debido a sus manifestaciones físicas, el requerimiento de cuidados constantes de la piel y el estigma social asociado. La visibilidad de su enfermedad puede conllevar dificultades psicosociales y problemas psicológicos, por lo que hay que abordar tanto las necesidades físicas como las emocionales.
Las asociaciones de pacientes juegan un papel crucial en la mejoría de la calidad de vida, proporcionando información, apoyo y recursos. En España, la Asociación Española de Ictiosis (ASIC) (http://www.ictiosis.org) se dedica a ayudar a personas afectadas y sus familias, promoviendo la conciencia, el apoyo psicosocial y la integración de los pacientes en la sociedad. ASIC también fomenta la investigación y colabora con profesionales de la salud para mejorar el manejo de la enfermedad. Esta labor está representada en Latinoamérica por distintas asociaciones en función de cada país, como http;//www.ictiosis.cl en Chile, entre otras.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



