

















La patología vascular oclusiva es causante de diversas y variadas manifestaciones clínicas, algunas de ellas con catastróficas consecuencias para el paciente. Dado que las causas de tal oclusión son muy variadas, hemos abordado en un artículo previo reciente en esta misma revista las causas trombóticas. En el presente artículo recopilamos diversas causas adicionales de oclusión intravascular.
Vascular occlusion has multiple, diverse clinical manifestations, some of which can have grave consequences for patients. It also has a wide variety of causes, including thrombi, which we recently addressed in partI of this review. In this second part, we look at additional causes of vascular occlusion.
En un artículo previo reciente en esta misma revista discutimos las principales causas de trombosis como formas de oclusión vascular. Esta segunda parte la dedicamos a una recopilación de causas de oclusión vascular variadas. En muchos de estos cuadros, no obstante, las manifestaciones clínicas son en muchas ocasiones similares, con signos diversos de isquemia, livedo reticularis, ulceraciones…, por lo que la histopatología desempeña un papel fundamental en la identificación
CoagulopatíasCoagulopatías sistémicasIncluimos en este epígrafe cuadros que cursan con defectos sistémicos de la coagulación, generalmente por alteración de alguna de las proteínas/factores que participan en ella.
Defectos de proteínas C y SDefinición. El sistema proteínaC-proteínaS es un importante inhibidor de la coagulación cuya acción es especialmente relevante en la microvasculatura. Defectos de dicho sistema pueden dar lugar a síndromes de oclusión microvascular graves que pueden terminar en una púrpura fulminante1,2.
Clínica. La presentación clínica varía según la severidad del déficit. Mutaciones homocigóticas condicionantes de una disfunción severa del sistema pueden dar lugar a cuadros graves de púrpura fulminante neonatal, con lesiones de púrpura retiforme y gangrena de extremidades1,3. Por el contrario, deficiencias parciales en estados de heterocigosis pueden pasar desapercibidas o ponerse de manifiesto tras un desencadenante externo (como el tratamiento con anticoagulantes orales)1,2.
Histopatología. Es similar a la de otros síndromes de oclusión microvascular no inflamatorios, con trombosis blanda de vasos de distinto tamaño en dermis e hipodermis y un mínimo o ausente infiltrado inflamatorio, sin signos de vasculitis.
Necrosis inducida por cumarina/warfarinaDefinición. El acenocumarol y la warfarina son anticoagulantes orales de uso muy extendido que actúan mediante la inhibición del metabolismo de la vitaminaK produciendo una disminución de los niveles plasmáticos de los factores de la coagulación vitaminaK-dependientes (II, VII, IX y X) y del sistema inhibitorio proteínaC-proteínaS4. Debido a la menor vida media de estos últimos, durante los primeros días de tratamiento se produce un estado protrombótico paradójico5,6. Más recientemente estos fármacos también han sido asociados a cuadros de calcifilaxis no urémica7.
Clínica. La necrosis cutánea asociada a los anti-vitaminaK se produce típicamente durante la primera semana de tratamiento, coincidiendo con el descenso relativo de los niveles de proteínasC yS4,5. Se desarrolla característicamente en zonas de importante panículo adiposo, como las mamas o los muslos. La clínica se superpone a la de otras entidades que cursan con púrpura retiforme, con lesiones eritematosas, hemorrágicas, que suelen tener un centro necrótico y que pueden presentar morfología ramificada en su periferia4,7. La calcifilaxis no urémica asociada a warfarina también se presenta como púrpura retiforme, pero aparece tras meses o años de tratamiento7.
Histopatología. La biopsia de lesiones de necrosis cutánea clásica asociada a anti-vitaminaK muestra hallazgos típicos de vasculopatía trombótica no inflamatoria4,5, con presencia de depósitos eosinófilos de fibrina, de aspecto amorfo, ocluyendo el interior de múltiples vasos4. El tamaño de los vasos ocluidos es variable, y pueden afectarse tanto vasos superficiales a nivel de la dermis papilar como otros más profundos, en la dermis reticular, o incluso en la unión dermohipodérmica4-6. Es frecuente la presencia de extravasación hemática que rodea a los vasos afectos y de necrosis epidérmica asociada4. El infiltrado inflamatorio acompañante es muy escaso o incluso ausente, y no se encuentran fenómenos de vasculitis4,5. En la calcifilaxis no urémica asociada a warfarina los hallazgos histopatológicos son superponibles a los de la calcifilaxis clásica7.
Coagulación intravascular diseminada (CID)Definición. Se trata de un síndrome adquirido que se caracteriza por la activación intravascular de la coagulación debida a diferentes causas. La activación de la coagulación genera fibrina que se deposita en los vasos sanguíneos con la consiguiente obstrucción de los mismos y el compromiso de la irrigación sanguínea de órganos y sistemas. A su vez se produce un consumo de las proteínas de la coagulación y las plaquetas, lo que ocasiona importantes hemorragias. Para diagnosticar una CID es necesario la existencia de una enfermedad de base y que se cumplan una serie de criterios analíticos. La causa más frecuente es la sepsis bacteriana8,9.
Clínica. La afectación es multiorgánica. Las lesiones cutáneas son con frecuencia el primer signo de la CID y suelen ser petequias y placas purpúricas que pueden evolucionar a ampollas hemorrágicas. Dos entidades que con mucha frecuencia (pero no siempre) se desarrollan en el seno de una CID son la púrpura fulminans y la gangrena simétrica periférica. La primera se caracteriza por lesiones purpúricas confluentes que evolucionan rápidamente a lesiones necróticas bien delimitadas, y su mortalidad alcanza el 50%. La segunda consiste en isquemia simétrica de áreas distales10,11 (fig. 1A).
. C: Lesión de pocos días de evolución en CID. Formación de ampolla subepidérmica con epidermis necrosada y trombos de fibrina en pequeños vasos sanguíneos dérmicos (H&E 200x).")
A: Lesión de gangrena simétrica periférica caracterizada por una placa isquémica bien delimitada en la mano izquierda con decoloración azulada de las uñas. B: Trombos de fibrina en vasos del plexo superficial y extravasación hemática dérmica (H&E 400x). C: Lesión de pocos días de evolución en CID. Formación de ampolla subepidérmica con epidermis necrosada y trombos de fibrina en pequeños vasos sanguíneos dérmicos (H&E 200x).
Histopatología. Los hallazgos histopatológicos de la CID no son patognomónicos, pero pueden ser muy orientativos en relación con un contexto clínico determinado. En las biopsias cutáneas se observan trombos fibrinoplaquetarios que afectan predominantemente a pequeños vasos sanguíneos de la dermis, en número variable, de escasos a múltiples. Con menor frecuencia se observa oclusión de pequeñas arterias en la dermis reticular. También es frecuente la existencia de extravasación hemática más prominente en los casos de púrpura fulminans (fig. 1B). El infiltrado inflamatorio que se observa suele ser mínimo o inexistente. Biopsias de lesiones de pocos días de evolución pueden mostrar necrosis epidérmica, ampollas subepidérmicas y necrosis de glándulas ecrinas y folículos pilosos (fig. 1C). Las lesiones clínicas que han progresado a gangrena mostrarán una extensa necrosis de todas las estructuras cutáneas10. Teniendo en cuenta que la causa más frecuente de la CID es la sepsis, no es infrecuente observar patrones mixtos de afectación de vasculopatía trombótica y de vasculopatía séptica con necrosis fibrinoide parietal vascular o invasión bacteriana parietal. En estos casos el infiltrado inflamatorio dérmico es más prominente12.
Síndrome antifosfolípido (SAF)Definición. Se trata de una vasculopatía trombótica, arterial o venosa, de naturaleza autoinmune, asociada a la presencia de autoanticuerpos antifosfolípidos (anticoagulante lúpico, anticardiolipina y anti-β2glucoproteína). Probablemente es la trombofilia adquirida más frecuente. Puede ser idiopática o estar asociada a distintas condiciones, entre las que se incluyen enfermedades autoinmunes (sobre todo lupus eritematoso [LE]), infecciones, neoplasias hematológicas o sólidas, fármacos y otras situaciones menos frecuentes13.
Clínica. Afecta con mayor frecuencia a mujeres jóvenes. Suele presentarse con necrosis cutánea, livedo reticularis, fenómeno de Raynaud o hemorragias subungueales como primera manifestación del síndrome, sobre todo en las formas primarias y las asociadas a LE. Para hacer el diagnóstico es necesario cumplir un criterio clínico junto con presencia de uno de los anticuerpos mencionados arriba14. Los criterios clínicos son: trombosis vascular confirmada en cualquier localización (piel, SNC, corazón, pulmón, riñón, digestivo) o historia de patología gestacional (muerte fetal intraútero, abortos espontáneos, nacido pretérmino). Tras el diagnóstico es aconsejable seguir al paciente durante 5-10años, ya que muchos evolucionan a un LE sistémico. En menos del 1% de casos se desarrolla un SAF catastrófico, con una mortalidad superior al 50%, secundario a una microangiopatía trombótica generalizada que afecta varios órganos simultáneamente o en un corto período de tiempo15.
Histopatología. Vasculopatía con trombos de fibrina, sin inflamación de la pared vascular, en vasos arteriales o venosos de cualquier calibre (fig. 2A,B). En ocasiones pueden observarse escasos linfocitos o células plasmáticas. En lesiones de larga evolución pueden existir fenómenos de recanalización y proliferación vascular (angioendoteliomatosis reactiva).
Coagulopatías vasculares. A. Trombosis venosa (H&E, 100x). B. Trombosis en arteriola (H&E, 200x).")
La coagulación puede ser defectuosa también por causas del continente (en este caso la pared vascular) que dispara de modo anormal el mecanismo de coagulación, por defectos de su estructura o composición (generalmente del endotelio).
Síndrome de Sneddon (SS)Definición. Se trata de un raro trastorno neurocutáneo episódico o crónico, lentamente progresivo, caracterizado por livedo racemosa generalizada y accidentes cerebrovasculares de repetición16. Se considera primario en más de la mitad de los casos, o secundario si coexisten enfermedad autoinmune o trombofilia (síndrome antifosfolípido). Se ha descrito además una forma familiar con mutación en CECR1(cat eye syndrome region, candidate1) que produce déficit de proteína ADA2 (adenosine deaminase2)17.
Clínica. Alrededor del 80% de los pacientes son de sexo femenino, con una edad media al diagnóstico de 40años, aunque los síntomas pueden iniciarse en edad pediátrica. La principal manifestación cutánea es la livedo racemosa, aunque en un primer momento se había descrito como livedo reticularis18. Se inicia normalmente en la mitad posterior e inferior del tronco y progresa al dorso de brazos y muslos, preservando los pies y la zona facial. Las lesiones son no dolorosas y raramente se acompañan de edema o se ulceran. Su severidad no se correlaciona con la de los síntomas neurológicos. Raramente se pueden encontrar acrocianosis, fenómeno de Raynaud, angiomatosis, úlceras cutáneas circunscritas, liquen plano anular atrófico y gangrena. Las manifestaciones neurológicas suelen comenzar con cefalea, mareo y vértigo, tras lo que suceden accidentes vasculares transitorios recurrentes y finalmente infartos cerebrales isquémicos o hemorrágicos. En esta fase se producen deterioro cognitivo progresivo y demencia.
Histopatología. El estudio histológico que ofrece mayor rentabilidad es el de muestras profundas obtenidas de la porción central y no del anillo de la livedo racemosa16. Las primeras lesiones, que raramente se biopsian, consisten en subendotelitis e inflamación mural de vasos arteriales de pequeño y mediano calibre. Lo más frecuente es observar engrosamiento subendotelial por proliferación de miocitos con oclusión total o subtotal de la luz vascular en dermis e hipodermis. En la fase final la hipoxia local estimula la proliferación endotelial y angiogénesis adventicial19.
Vasculopatía livedoide (VL)Definición. Es una frecuente enfermedad vascular cutánea trombótica crónica que cursa con isquemia y que tiene como causas más importantes las coagulopatías, las enfermedades autoinmunes y ciertas infecciones, a lo que se añade la insuficiencia venosa periférica como factor muy relevante facilitador de trombosis.
Clínica. La VL es más propia de mujeres de edad media que desarrollan en la piel de las extremidades inferiores pápulas y placas más o menos purpúricas, simétricas, dolorosas y persistentes, que dan lugar a úlceras de evolución tórpida y que, según el tamaño lesional, pueden curar u originar una progresiva característica atrofia en porcelana de la piel (llamada atrofia blanca). La causa de la trombosis son estados de hipercoagulabilidad sanguínea debidos a factores procoagulantes circulantes del tipo de anticuerpos (crioglobulinas, anticuerpos antifosfolípidos idiopáticos o surgidos en el contexto de enfermedades autoinmunes como el lupus eritematoso, anticuerpos generados en la hepatitis viral, etc.), trombocitosis o anomalías hereditarias de los factores de la coagulación (por ejemplo, mutación del factor V Leiden, déficit de factores fibrinolíticos, etc.). A esto se suman la insuficiencia venosa crónica periférica y los traumatismos como factores facilitadores de la trombosis. En muchos casos la enfermedad es idiopática. En la piel se manifiesta como pápulas y placas purpúricas que tienden a ulcerarse originando cicatrices de aspecto porcelánico (fig. 3A).
. C: Foto de detalle de la anterior en la que se aprecia engrosamiento de la pared vascular, incremento del número de pericitos y depósitos de material fibrinoide eosinófilo y homogéneo. En torno al vaso se observa hemorragia, ocasionales macrófagos con hemosiderina, infiltrado linfomacrofágico y fibrosis colagénica perivascular (H&E 200x).")
Vasculopatía livedoide. A: Mujer de 62 años afecta de lupus eritematoso e insuficiencia venosa que muestra en la extremidad inferior eritema, múltiples ulceraciones y discromía de la piel por hemorragia. B: Piel que muestra el desarrollo de agregados vasculares de carácter glomeruloide en la dermis superficial (H&E 100x). C: Foto de detalle de la anterior en la que se aprecia engrosamiento de la pared vascular, incremento del número de pericitos y depósitos de material fibrinoide eosinófilo y homogéneo. En torno al vaso se observa hemorragia, ocasionales macrófagos con hemosiderina, infiltrado linfomacrofágico y fibrosis colagénica perivascular (H&E 200x).
Histopatología. La biopsia cutánea debe incluir piel adyacente al área ulcerada para que esta sea demostrativa. La histopatología de la VL no es patognomónica. En la VL se afectan los vasos de la dermis superficial y media conformando agregados de vasos dilatados que adquieren un aspecto glomeruloide, con depósito de material hialino fibrinoide PAS positivo en la pared vascular, trombosis y hemorragia, que no debe confundirse con vasculitis. Además, se aprecia un variable infiltrado linfomacrofágico perivascular. Las úlceras dan lugar a fenómenos de cicatrización. En los casos más evolucionados surge la atrofia epidérmica y una fibrosis colagénica intensa por isquemia crónica, que es la razón de la característica atrofia en porcelana apreciable clínicamente (fig. 3B,C). En el estudio de inmunofluorescencia directa se pueden encontrar depósitos de inmunoglobulinas, complemento y fibrinógeno, lo que sugiere una naturaleza inmune del proceso. Para el diagnóstico diferencial con otras vasculopatías trombosantes es importante la correlación clinicopatológica.
Histopatológicamente se diferencia de la crioglobulinemia tipoI y de la microangiopatía diabética en que estas no muestran arquitectura glomeruloide de los vasos. Es complicada la diferenciación histopatológica respecto a la dermatitis de estasis y la acroangiodermatitis, pero estas en general presentan marcados depósitos férricos o una intensa proliferación vascular, respectivamente.
Enfermedad de DegosDefinición. La enfermedad de Degos, también llamada enfermedad de Kohlmeier-Degos o papulosis atrófica, es una enfermedad rara, de la que hay menos de 200casos descritos20. Afecta fundamentalmente a mujeres de entre 20 y 50años, aunque se han descrito casos pediátricos21. Su etiopatogenia es desconocida; se ha relacionado con coagulopatías, vasculitis y alteraciones del revestimiento celular endotelial, posiblemente en relación con factores del complemento20-22. La existencia de casos familiares ha sugerido una posible predisposición genética.
Clínica. Presenta lesiones cutáneas que son consideradas como patognomónicas: pápulas atróficas con centro blanquecino porcelánico y halo eritematoso telangiectásico de tamaño menor a 1cm21 (fig. 4A). Con dermatoscopia producen un patrón característico en «corona de espinas»23. Las lesiones afectan al tronco y a las extremidades, respetando el cuero cabelludo y la cara. Actualmente se plantea dividir la enfermedad de Degos en dos formas: una benigna, con afectación exclusivamente cutánea y buen pronóstico (71% de los casos), y una forma sistémica (papulosis atrófica maligna), que puede ocurrir simultánea o posteriormente a la afectación cutánea. La forma sistémica puede producir complicaciones graves e incluso la muerte en el 75% de los casos por afectación intestinal (perforación), cerebral, pulmonar, renal, etc.20,21.
. C: Detalle del vaso mencionado en B, con numerosos trombos intravasculares (H&E 400x).")
Enfermedad de Degos. A: Las pápulas atróficas con centro blanquecino porcelánico y halo eritematoso telangiectásico se consideran un hallazgo patognomónico de la enfermedad. B: Infarto en forma de cuña con el vértice hacia abajo. En la punta del vértice se observa un vaso con discreto infiltrado linfocitario crónico perivascular (H&E 20x). C: Detalle del vaso mencionado en B, con numerosos trombos intravasculares (H&E 400x).
Histopatología. Los hallazgos dermatopatológicos dependen del tiempo de evolución de la enfermedad. En lesiones establecidas se observa característicamente un infarto en cuña de la dermis con lesión vascular (fig. 4B), identificando tumefacción endotelial y trombos intraluminales de fibrina (fig. 4C). La dermis cercana muestra agregados inflamatorios linfocitarios perivasculares y perianexiales con depósito intersticial de mucina. La epidermis puede mostrar daño isquémico, atrofia, lesión de la interfase e hiperqueratosis24. Las lesiones tempranas pueden ser muy similares a las del lupus eritematoso cutáneo, con agregados linfocitarios perivasculares y depósito intersticial de mucina25.
Síndromes de oclusión por hematíesEn algunos cuadros, las células que se agregan y que son las principales responsables de la oclusión vascular son los hematíes.
Adhesión reticulocitaria por estrésDefinición. La adhesión reticulocitaria por estrés se debe a la alteración de las propiedades reológicas de los eritrocitos, que cambian su viscosidad y aumentan su propensión a producir trombos26. Aunque hay distintas enfermedades que alteran las propiedades reológicas de los eritrocitos, este término se aplica principalmente a la anemia falciforme, en la que hay un doble mecanismo implicado en la oclusión intraluminal. Por un lado se produce liberación de reticulocitos de estrés CD36+, con mayor tendencia a adherirse al endotelio, que, además, aumenta su expresión de ICAM-1, una molécula de adhesión. Ambos procesos conllevan una activación de la vía de la coagulación, y la hipoxia resultante incrementa tanto la liberación de nuevos reticulocitos de estrés como la expresión endotelial de ICAM-1.
Clínica. A nivel cutáneo se observan úlceras muy dolorosas, únicas o múltiples, con predilección por la zona perimaleolar, sobre todo interna27. No obstante, estos pacientes pueden tener además ictus, hipertensión pulmonar, infartos esplénicos y alteración de la función renal secundaria a fenómenos trombóticos en diversos órganos.
Histopatología. Se observa habitualmente ulceración de la epidermis con tejido de granulación, además de presencia de trombos de fibrina en los vasos, que pueden presentar hialinización de su pared, similar a lo observado en la vasculopatía livedoide28. La epidermis adyacente a la úlcera se caracteriza habitualmente por acantosis e hiperqueratosis.
ÉmbolosUn émbolo es cualquier estructura que, arrastrada por la sangre, acaba produciendo la obstrucción parcial o total de un vaso. El émbolo puede ser el propio trombo que, después de haberse formado in situ, se ha desprendido hasta otro punto lejano, pero también puede estar hecho por materiales ajenos a la sangre e incluso ajenos totalmente al cuerpo humano.
Material endógenoColesterolDefinición. Es una enfermedad multisistémica que se produce por la liberación de cristales de colesterol hacia la circulación tras la rotura de una o más placas ateroscleróticas. Estos cristales migran en dirección distal, ocluyendo parcial o totalmente arterias de mediano y pequeño calibre de diversos órganos, entre los que destaca la piel. Aun cuando puede ocurrir de forma espontánea, generalmente va precedida de procedimientos angioinvasivos o del uso de anticoagulantes o fibrinolíticos que desestabilizan la placa29.
Clínica. Es muy variada, y lo más frecuente es la presencia de livedo reticularis, pápulas y nódulos purpúricos, petequias y cianosis que afectan la región distal de las extremidades (fig. 5A). En casos más severos es posible observar úlceras y gangrena.
. C: Émbolos por cristales de colesterol. En la fase tardía puede haber fibrosis alrededor de los cristales, con oclusión completa de la luz del vaso sanguíneo (H&E 200x).")
Émbolos por cristales de colesterol. Livedo reticularis planta pie derecho. B: Émbolos por cristales de colesterol. Espacios aciculares vacíos en el interior de un vaso sanguíneo. Dichos espacios contenían los cristales de colesterol que quedan disueltos durante el procesamiento de la biopsia. Hay respuesta de células gigantes multinucleadas rodeando los espacios aciculares vacíos (H&E 200x). C: Émbolos por cristales de colesterol. En la fase tardía puede haber fibrosis alrededor de los cristales, con oclusión completa de la luz del vaso sanguíneo (H&E 200x).
Histopatología. El hallazgo más característico y específico es la presencia de hendiduras alargadas, biconvexas y en forma de aguja en el interior de uno o más vasos de mediano y pequeño calibre (100-200μm) (fig. 5B). Estas estructuras se corresponden a la imagen en negativo de los cristales de colesterol disueltos durante el procesamiento de la muestra y se suelen observar en la luz de los vasos de la unión dermohipodérmica30. En ocasiones, sin embargo, para lograr su visualización puede ser necesaria la realización de múltiples secciones histológicas. Otros hallazgos frecuentes que acompañan y rodean a los cristales son la presencia de neutrófilos y eosinófilos en fases iniciales y de células multinucleadas y fibrosis en fases tardías16 (fig. 5C).
OxalatoDefinición. El depósito de oxalato se produce fundamentalmente en las hiperoxalurias primarias, un grupo de trastornos metabólicos de herencia autosómica recesiva que cursan con superproducción de oxalato sérico y el subsecuente depósito en los tejidos. Las oxalurias secundarias se producen por ingesta o aporte excesivos de oxalato o sus precursores (envenenamiento por etilenglicol, anestesia por metoxiflurano, ingesta excesiva de ácido ascórbico, diversas enfermedades intestinales, resección ileal, hemodiálisis crónica…).
Clínica. Los síntomas suelen ser a edad temprana y fundamentalmente renales, por urolitiasis recurrente y fallo renal. En la piel, el depósito intravascular de oxalato produce livedo reticularis, acrocianosis, gangrena periférica y ulceraciones31.
Histopatología. Vasos de mediano tamaño ocluidos por un material cristalino marronáceo. Los cristales birrefringen con luz polarizada mostrando formas aciculares y rectangulares. Puede acompañarse de necrosis de la dermis y la hipodermis32.
Vasculopatía por cristales de globulina (cristalglobulinemia)Definición. La cristalglobulinemia consiste en la cristalización irreversible de proteínas monoclonales en el interior de los vasos en pacientes con mieloma múltiple o gammapatía de significado incierto33. Aunque se han descrito menos de 30 casos en la literatura, probablemente no es tan infrecuente como se piensa. En los casos en que la proteína monoclonal se cristaliza como consecuencia de la crioprecipitación, la enfermedad recibe el nombre de «criocristalglobulinemia». Los cristales precipitados producen daño endotelial con activación de la cascada de la coagulación, trombosis, fenómenos oclusivos y daño isquémico subsecuente.
Clínica. Produce en la piel lesiones ulceradas y purpúricas, más frecuentes en extremidades distales (fig. 6A). Puede afectar al riñón con alteración severa de la función renal y trombosis de la arteria renal bilateral. También puede afectar a otros órganos, produciendo neuropatía periférica, poliartropatía, etc.34.
.")
Histopatología. Histológicamente se observa una vasculopatía trombótica producida por estructuras cristaloides intraluminales localizadas en vasos de pequeño y mediano calibre (fig. 6B). Los cristales son PAS positivos, no birrefringentes con luz polarizada, y se rodean de fibrina y hematíes. Están formados por inmunoglobulinas monoclonales que pueden demostrarse por inmunofluorescencia. La morfología de los cristales es muy variable: pueden formar agujas, estructuras romboidales, rectangulares o cuboideas. No existe relación entre la forma del cristal, el tipo de inmunoglobulina y las manifestaciones clínicas35,36.
TumoresMixoma auricularDefinición. El mixoma, aunque raro, es el tumor cardiaco más frecuente. En el 75% de los casos la lesión está situada en la aurícula izquierda. Es más frecuente en mujeres de entre 30 y 60años37. El 90% de los casos son esporádicos, y el resto forma parte del complejo de Carney u otras enfermedades hereditarias con origen en la mutación del gen PRKAR1A16. El mixoma cardiaco es difícil de sospechar, ya que produce síntomas constitucionales leves y resultados inespecíficos en las pruebas habituales. En ocasiones (30-40%) produce émbolos de fragmentos del tumor que afectan preferentemente al sistema nervioso central y a la piel37-39.
Clínica. En la piel, la clínica típica son episodios recurrentes de lesiones transitorias, que suelen ser dolorosas, situadas en las extremidades. Las lesiones son máculas o pápulas eritematosas más o menos purpúricas (fig. 7A). Otras alteraciones descritas son livedo reticularis, cianosis digital, petequias, necrosis o úlceras acrales, hemorragias subungueales «en astilla», lesiones anulares o serpiginosas, y entumecimiento de manos o pies16,37-40. La biopsia de piel puede ser fundamental, ya que a veces los émbolos cutáneos son la primera o única manifestación de la enfermedad. Dado que las alteraciones se suelen producir dentro de las arteriolas de la dermis profunda o hipodermis, es importante que la biopsia sea profunda.
.")
Histopatología. La biopsia suele ser normal o presentar alteraciones inespecíficas, como fibrina intravascular. Solo en casos excepcionales la luz de alguna arteriola aparece ocluida por un tejido laxo mixoide muy vascularizado en el que hay células estrelladas o fusiformes (fig. 7B). La lesión resalta con las técnicas de hierro coloidal y azul alcián16,37-39.
Linfoma B de célula grande intravascular (LBCG-IV)Definición. El LBCG-IV es una entidad muy poco frecuente definida por la proliferación de célulasB grandes neoplásicas de forma predominante o exclusiva dentro de vasos, especialmente capilares41.
Clínica. La edad media de los pacientes al momento del diagnóstico es de 70años y el espectro de presentación clínica es heterogéneo. Se han descrito tres variantes clínicas: la clásica, caracterizada por fiebre y síntomasB asociados a disminución del estado funcional, lesiones cutáneas, alteración del nivel de consciencia o hipoxemia de causa no aclarada; la variante cutánea, que se presenta como lesiones tipo erupciones maculopapulares, púrpura palpable, piel de naranja, celulitis, nódulos eritematovioláceos algunos ulcerados, en ausencia de compromiso sistémico; y la variante asociada a síndrome hemofagocítico42,43.
Histopatología. El LBCG-IV se caracteriza por una proliferación de células grandes, de morfología variable (centroblastos, inmunoblastos o plasmablastos), con un patrón de crecimiento intravascular que puede ser discohesivo (células aisladas intraluminales), cohesivo (células que ocluyen la luz vascular) o marginal (adheridas al endotelio). Las células atípicas son positivas para CD20 mayoritariamente con un fenotipo no centrogerminal (CD10−, BCL6+/−), con expresión de MUM-1 (fig. 8). La coexpresión de CD5 se ha informado en un porcentaje cercano al 40% de los casos y el índice proliferativo medido mediante la expresión de ki-67 suele ser elevado (aprox. 90%)44,45. Un punto importante de discusión es el papel de la biopsia cutánea sobre piel sin lesión para el diagnóstico del LBCG-IV. A pesar de que en la literatura no hay consenso con respecto a su utilidad, se ha informado una sensibilidad cercana al 80% y una especificidad cercana al 100% en biopsias incisionales con representación de panículo adiposo46. Dentro de los diagnósticos diferenciales del LBCG-IV en biopsia cutánea las proliferaciones de célulasT atípicas CD30+ requieren especial atención. Este tipo de pseudolinfomas se han descrito en relación con enfermedades inflamatorias, tumores o trauma47,48.
Angiosarcoma. Positividad inmunohistoquímica para CD20 (B) y MUM-1 (C) (ambas 400x). El índice proliferativo (D)(Ki-67) es alto (400x).")
Definición. El angiosarcoma es un tumor maligno poco frecuente que deriva del endotelio vascular, de comportamiento agresivo local y con capacidad metastatizante49. Dada la posibilidad de crecimiento intravascular, puede generar oclusión y embolia16.
Clínica. En la piel se clasifica como primario o secundario, estando este último relacionado con radioterapia o con linfedema crónico49. En general, cursan con edema (facial en el caso del angiosarcoma de Wilson Jones) y con áreas contusiformes o congestivas50. Cuando genera oclusión vascular, clínicamente puede cursar con lesiones retiformes, tipo livedo, simulando vasculitis (fig. 9A), púrpura o incluso úlceras e infartos16.
.")
Histopatología. El patrón histológico es muy variable. En casos bien diferenciados se aprecia proliferación vascular infiltrativa con endotelios levemente atípicos, mientras que en casos menos diferenciados muestra patrón más sólido, fusiforme o epitelioide50. En las variantes sólidas resulta de utilidad la presencia de vacuolas intracitoplasmáticas (luz vascular rudimentaria), hematíes e infiltrado inflamatorio parcheado51.
Algunas variantes pueden mostrar crecimiento intravascular con aspecto de linfangitis carcinomatosa (fig. 9B). Esto ocurre con la variante epitelioide, así como con la embolización de angiosarcomas de grandes vasos52. Será de utilidad demostrar el inmunofenotipo de las células tumorales con marcadores vasculares (CD31, CD34, ERG), incluyendo en muchas ocasiones los de diferenciación linfática (podoplanina, D2-40, LYVE-1 y PROX-1)50,51. Ha de recordarse que en algunos casos indiferenciados disminuye la expresión de marcadores vasculares y pueden adquirir marcadores epiteliales, tales como MNF116 y citoqueratina AE1/AE316,51. En este sentido, la morfología epitelioide endoluminal plantea el diagnóstico diferencial con el hemangioendotelioma epitelioide, que tiene la translocación característica del WWTR1-CAMTA1 o del YAP1-TFE353.
Metástasis intravascularesDefinición. El término «metástasis cutánea intravascular» hace referencia a la diseminación de un tumor primario bien a través de los vasos linfáticos o bien a través de los vasos hemáticos54. La diseminación intralinfática es mucho más frecuente y puede encontrarse descrita como linfangitis carcinomatosa, carcinoma inflamatorio o carcinoma erisipeloide55. En este artículo se estudia la oclusión intravascular luminal hemática, por lo que nos referiremos a las metástasis intravasculares hemáticas, también denominadas carcinoma telangiectásico56. Se ha demostrado que las metástasis hematógenas se originan mediante la fijación de las células tumorales al endotelio vascular. Tras esta fijación las células proliferan originando la metástasis, sin necesidad de extravasarse al intersticio57.
Clínica. Se presenta como una placa eritematosa e infiltrada con telangiectasias evidentes en superficie (fig. 10). En otras ocasiones adopta una imagen pseudovesicular que recuerda a un linfangioma circunscrito. Las lesiones clínicamente asemejan un angiosarcoma, especialmente cuando se localizan en la extremidad cefálica o en la región mamaria58.

Histopatología. Se aprecian acúmulos de células neoplásicas junto a eritrocitos ocupando la luz dilatada de los vasos de la dermis. No se encuentran células malignas entre los haces de colágeno ni formando acúmulos por fuera de los vasos. Mediante doble tinción con CD31 y D2-40 puede demostrarse que los agregados tumorales ocupan tan solo los vasos sanguíneos caracterizados por ser CD31 positivos y D2-40 negativos (fig. 11).
.")
Trombos tumorales. Vasos dilatados en la totalidad de la dermis, con la luz totalmente ocupada por células pleomórficas de apariencia epitelial entremezcladas con hematíes. La inmunotinción con CD 31 demuestra la naturaleza intravascular de la proliferación tumoral (A: H&E 20x; B: H&E 400x; C: H&E 200x; D: CD31 100x).
Definición. El embolismo cutáneo no está restringido a material endógeno, sino que más raramente puede corresponder a material exógeno que se ha desprendido durante procedimientos endovasculares o se ha empleado en el tratamiento quimioembólico de algunos tumores. Entre los materiales extraños que pueden ser causa de émbolos cutáneos se encuentran los revestimientos hidrofílicos o hidrófobos presentes en los dispositivos intravasculares, cuya misión es reducir la fricción causada al insertar dicho dispositivo en los vasos, minimizando el daño endotelial, y para reducir la incidencia de espasmos arteriales59. Aunque su presencia en la piel es relativamente rara, se ha reseñado con frecuencia creciente debido al empleo cada vez más generalizado de múltiples técnicas intravasculares tanto diagnósticas como terapéuticas.
Clínica. Clínicamente, la manifestación más habitual es la livedo reticularis, entre cuyas múltiples etiologías descritas debe incluirse esta posibilidad. También se han descrito casos que se han presentado con lesiones purpúricas retiformes (fig. 12), úlceras o gangrena59-61.

Histopatología. El estudio histológico revela la oclusión de los vasos dérmicos o del tejido celular subcutáneo por material basófilo, amorfo o granular, que en muchos casos presenta una característica disposición laminar (fig. 13). Muestra positividad con hierro coloidal o azul alcián y no brilla con la luz polarizada. Es característica la ausencia de signos inflamatorios en la pared de los vasos afectos, aunque puede apreciarse cierto grado de inflamación inespecífica en la dermis adyacente60-62. Desde el punto de vista histopatológico, el diagnóstico diferencial más importante es el embolismo cutáneo secundario al mixoma auricular, que se caracteriza por la acumulación intravascular de material mixoide en las arteriolas dérmicas. Sin embargo, los fibroblastos estrellados dentro del estroma del émbolo, que imitan la estructura del tumor original, permiten el diagnóstico diferencial en la mayor parte de los casos y, si es necesario, la tinción inmunohistoquímica para marcadores de células endoteliales soluciona el problema16. Resulta muy difícil de diferenciar de otra complicación embólica cutánea mucho más infrecuente, como es el embolismo de ácido hialurónico tras procedimientos cosméticos63,64 o la inyección intraarticular en el tratamiento de la artrosis65,66. El antecedente clínico de uno u otro procedimiento es de gran importancia.
 es prácticamente indistinguible del ácido hialurónico (B). La disposición laminar del primero en algunos casos es la única diferencia.")
También se han reseñado casos de embolización cutánea de perlas liberadoras de fármacos con doxorubicina67. En la biopsia se aprecia material intravascular exógeno de color púrpura con morfología esférica, en algunos casos con extrusión focal en la dermis adyacente. Los principales diagnósticos diferenciales son la crioglobulinemia monoclonal y las micosis profundas, pero rara vez deben plantear problemas serios debido a su perfecta forma esférica. Excepcionalmente, una técnica simple como la tinción de PAS puede ser útil, ya que dichas esferas no se tiñen, al contrario que los trombos de la crioglobulinemia y los hongos16,67. También es esperable el incremento de este tipo de complicaciones con el uso cada vez más frecuente de este tipo de tratamiento.
Por último, hay referencias aisladas a otras complicaciones embólicas cutáneas en el contexto de la adicción a drogas por vía parenteral derivadas de la inyección de medicación oral machacada68 o del embolismo arterial terapéutico69.
MisceláneaIncluimos por último en este grupo una serie de causas de oclusión vascular cuya patogenia no se ajusta exactamente a ninguno de los grupos anteriores, bien por ser multifactorial o bien por ser distinta a los mecanismos explicados.
CalcifilaxisDefinición. La calcifilaxis (o arteriopatía urémica calcificante) es una complicación rara y severa de etiopatogenia desconocida, caracterizada por calcificación de la media de arteriolas y capilares en la dermis y el tejido celular subcutáneo. Es característica de pacientes con insuficiencia renal crónica en diálisis e hiperparatiroidismo, aunque también está descrita en pacientes con función renal normal70. Con mayor frecuencia afecta a pacientes de edad media-avanzada, de sexo femenino, raza blanca, diabéticos y VIH positivos. Se asocia con una alta tasa de morbimortalidad.
Clínica. Lesiones iniciales en miembros inferiores eritematosas de tipo livedo reticularis que progresan a lesiones de color violáceo muy dolorosas que forman placas o nódulos, afectan al tejido celular subcutáneo, se cronifican, ulceran he incluso presentan necrosis (fig. 14A). Se han descrito varios patrones de afectación desde el punto de vista clínico proximal y distal, siendo el proximal de peor pronóstico71.
. C: Detalle de los vasos trombosados (H&E 200x).")
Histopatología. El hallazgo más frecuente y característico es la calcificación de la media de arterias y arteriolas (fig. 15A,B) acompañada de necrosis y trombos intravasculares72 (fig. 14B,C). Se puede identificar proliferación intimal con estrechamiento de la luz de los vasos (fig. 15C,D). La calcificación intersticial es un hallazgo poco frecuente (fig. 15D). Se ha descrito calcificación periecrina en algunos casos. Excepcionalmente se han descrito cambios de tipo pseudoxantoma elástico73. También es habitual encontrar zonas de hemorragia y necrosis grasa en el tejido celular subcutáneo acompañada de un infiltrado inflamatorio linfohistiocítico afectando al lobulillo.
Hidroxiurea C: Calcificación y necrosis intersticial (H&E 400x). D: Proliferación intimal acompañada de calcificación de la pared del vaso (H&E 200x).")
Definición. El uso de hidroxiurea (agente no alquilante que bloquea el ciclo celular en la faseS) durante al menos un año y con dosis acumuladas de al menos 510g es capaz de producir alteraciones reológicas en los glóbulos rojos que pueden desencadenar el inicio de la cascada de la coagulación74,75.
Clínica. Son muy características las úlceras en la zona de maléolo, muy dolorosas y similares a las que se observan en la vasculopatía livedoide, y que al curar pueden dejar como secuela cicatrices blanquecinas estrelladas75.
Histopatología. Hallazgos inespecíficos; predomina la fibrosis dérmica (al ser un proceso crónico) y pueden identificarse algunos vasos con trombos de fibrina. Es casi más característico encontrar una dermatitis de interfase asociada a atrofia epidérmica con queratinocitos apoptóticos ocasionales y hemosiderófagos75.
Vasculopatía trombótica inducida por cocaína-levamisolDefinición. Desde 2005, la cocaína se adultera frecuentemente con levamisol (70-90% en EE.UU.), un polvo blanco de bajo coste que aumenta su volumen y también los efectos estimulantes de la cocaína al aumentar la dopamina cerebral76. El resultado es un producto con efectos trombóticos secundarios indeseables.
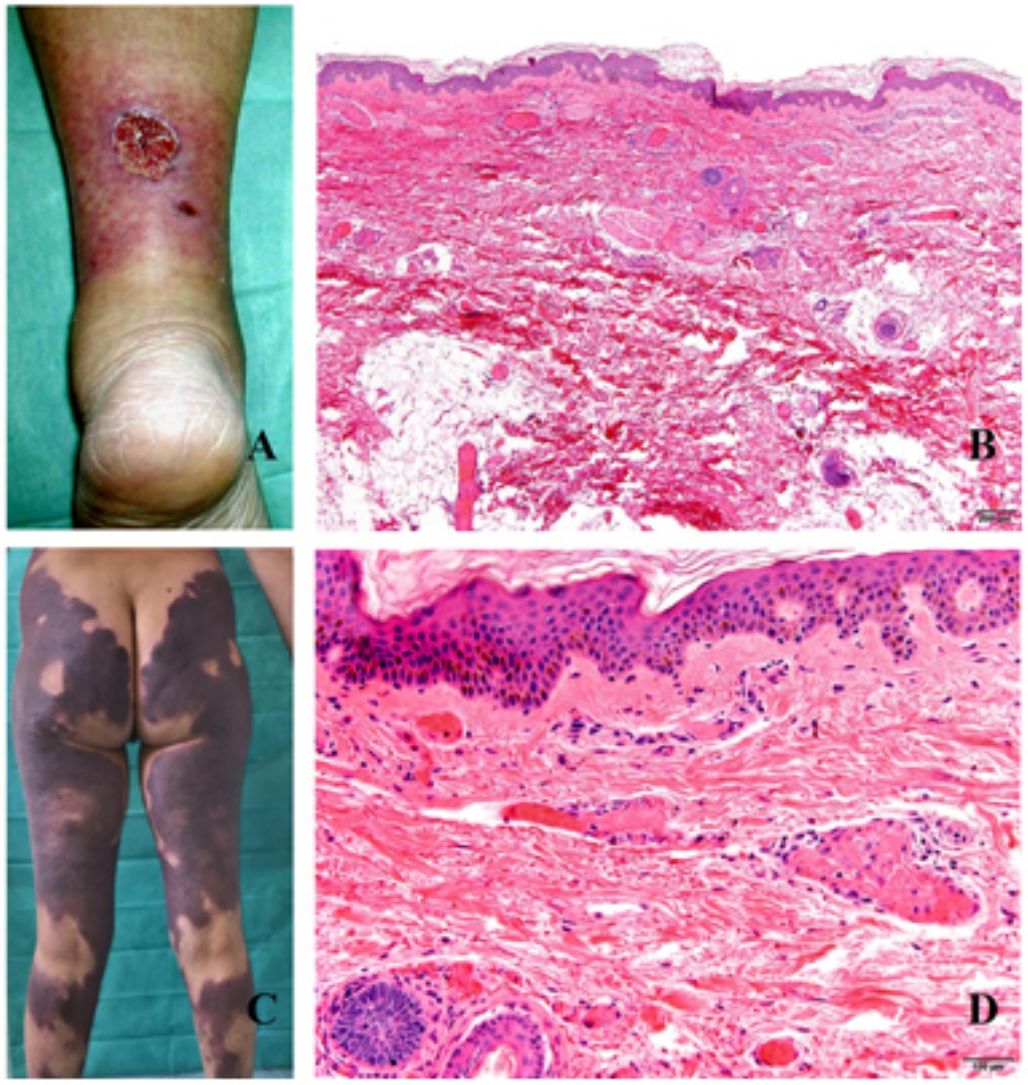
Clínica. En el síndrome de vasculopatía asociada a cocaína-levamisol la afectación cutánea es la manifestación clínica más frecuente. Se presenta con lesiones muy dolorosas que van desde la púrpura retiforme hasta placas purpúricas confluentes en más del 80% de casos (fig. 16A,C). Se pueden acompañar de necrosis y ampollas hemorrágicas. Dichas lesiones afectan principalmente a las extremidades superiores e inferiores, a cara y a abdomen, aunque tienen especial predilección por las orejas (>70%)77.
. B, Vasculopatía oclusiva asociada a cocaína-levamisol, con trombos de fibrina intravascular, sin signos de vasculitis, con extravasación hemática y sin necrosis epidérmica. H&E 40x. C: Síndrome de vasculopatía asociada a cocaína-levamisol: Lesiones purpúricas retiformes confluentes que afectan extensamente los miembros inferiores y glúteos. D: Síndrome de vasculopatía asociada a cocaína-levamisol: Vasculopatía oclusiva con trombos de fibrina intravascular, sin signos de vasculitis, con extravasación hemática y sin necrosis epidérmica. H&E 100x.")
A: Úlcera secundaria al tratamiento con hidroxiurea (Cortesía del Dr. Fernando Cabo, Servicio de Dermatología, Hospital Universitario de Ourense, España). B, Vasculopatía oclusiva asociada a cocaína-levamisol, con trombos de fibrina intravascular, sin signos de vasculitis, con extravasación hemática y sin necrosis epidérmica. H&E 40x. C: Síndrome de vasculopatía asociada a cocaína-levamisol: Lesiones purpúricas retiformes confluentes que afectan extensamente los miembros inferiores y glúteos. D: Síndrome de vasculopatía asociada a cocaína-levamisol: Vasculopatía oclusiva con trombos de fibrina intravascular, sin signos de vasculitis, con extravasación hemática y sin necrosis epidérmica. H&E 100x.
Histopatología. La histología es variada, pudiendo mostrar desde vasculitis leucocitoclástica y vasculitis trombótica a vasculopatía oclusiva con trombos de fibrina intravascular, sin vasculitis verdadera (fig. 16B, D). Las úlceras cutáneas son generalmente consecuencia de un grado avanzado de isquemia.
Este artículo es una iniciativa del Grupo Español de Dermatopatología de la Academia Española de Dermatología y Venereología. Todos los autores firmantes han contribuido por igual en la elaboración del manuscrito. El orden de autores se ha establecido por mero orden alfabético de apellidos.


