









Las displasias pilosas corresponden a alteraciones en la estructura del tallo del cabello y pueden ser congénitas o adquiridas. Se clasifican en dos grandes grupos según la presencia o no de fragilidad capilar. En la mayoría de los casos la valoración del paciente, la anamnesis y la exploración física nos llevarán al diagnóstico. En los últimos años, el uso de la tricoscopia se ha posicionado como una técnica útil y coste efectiva, sobre todo en niños, ya que permite lograr una adecuada exploración sin tener que arrancar los cabellos.
En algunas ocasiones las alteraciones en la estructura del cabello serán la clave diagnóstica de enfermedades más complejas, en las que la instauración de un tratamiento precoz puede mejorar el pronóstico.
El propósito de esta revisión es aportar las claves que permitan diagnosticar las displasias pilosas más frecuentes y valorar las alternativas terapéuticas disponibles en la actualidad.
Hair shaft disorders, involving dysplastic abnormalities in the shaft, may be either congenital or acquired. Two large categories have been defined according to the presence or not of hair fragility. A diagnosis can usually be made after taking a thorough medical history and performing a physical examination. Trichoscopy has become a useful, cost-effective tool in recent years, particularly for examining the hair of children, because it facilitates inspection without removal of hairs. Structural abnormalities in the hair shaft are sometimes clues to the diagnosis of more complex diseases in which early treatment can improve prognosis. This review describes key features that enable the diagnosis of the most common hair shaft disorders and discusses the various treatments currently available.

El tallo piloso es una estructura compuesta por una corteza rodeada por una cutícula protectora y, en el caso de pelos terminales, además es posible distinguir una médula central (fig. 1a). Esto le confiere propiedades únicas en cuanto a fuerza, flexibilidad y resistencia al medio ambiente1.
 Pelo terminal con médula en su interior. B) Moniletrix. C) Pseudomoniletrix. D) Pili torti. E) Tricorrexis invaginata. F) Tricotiodistrofia. G) Tricorrexis nodosa. H) Cabello burbuja. I) Cabello en anágeno suelto.")
Las displasias pilosas constituyen alteraciones en la estructura del tallo, que pueden estar provocadas por factores ambientales o por mutaciones genéticas2,3. Los pacientes, o sus padres suelen consultar por cambios en la apariencia y textura del cabello, así como por un aumento en la fragilidad o su falta de crecimiento1. El objetivo de esta revisión es presentar las claves diagnósticas que permitan reconocer las principales displasias pilosas, evaluar los factores desencadenantes, el pronóstico y las alternativas de tratamiento disponibles en la actualidad.
Durante la anamnesis de un paciente que consulta por una alteración en el cabello, debemos hacer preguntas dirigidas como: ¿cuándo comenzó el problema?, ¿está presente desde el nacimiento o es algo adquirido?, ¿existen alteraciones dentarias o en las uñas?, ¿hay otros familiares con hallazgos similares?, ¿qué hábitos de higiene o uso de productos cosméticos tiene? Es relevante averiguar la frecuencia del lavado del cabello, las técnicas de peinado, el uso de fijadores, alisadores permanentes, plancha de pelo o secador1,4.
A la hora de explorar el cabello, debemos recordar algunos puntos importantes1:
- •
Evaluar el aspecto general, el brillo, el rizado y el color del pelo.
- •
Valorar la distribución y verificar si la anormalidad es focal o difusa.
- •
Hacer el pull test (maniobra de Sabouraud o signo del arrancamiento), en el que se sujetan mechones de entre 20 y 60 cabellos entre los dedos índice y pulgar, y se traccionan con suavidad en distintas áreas del cuero cabelludo. El test es positivo si se desprenden más del 10% de los cabellos.
- •
Realizar el signo de la tracción o tugtest, que consiste en sujetar entre los dedos un mechón de cabellos a varios centímetros de la raíz, y estirar para detectar áreas de fragilidad.
- •
Explorar el cuero cabelludo.
- •
Utilizar pruebas complementarias no invasivas, como la fotografía y la tricoscopia, que permiten complementar la exploración física en la consulta.
- •
Usar pruebas semi-invasivas como el tricograma, la microscopía óptica y la microscopía electrónica en casos seleccionados5.
- •
Valorar la necesidad de biopsia del cuero cabelludo en casos seleccionados.
La clasificación de las displasias pilosas puede ser difícil, porque en ocasiones se superponen los hallazgos clínicos entre sí. Las displasias del cabello se agrupan según la presencia o ausencia de fragilidad en el tallo piloso (fig. 2)6. Una adecuada anamnesis junto a una minuciosa exploración del cabello, ayudándose de técnicas como la tricoscopia, la microscopia óptica (MO) o la microscopía electrónica (ME), nos permitirán establecer el diagnóstico (tabla 1).

Aproximación diagnóstica a las displasias pilosas.
Modificado de Mirmirani et al.1.
Clasificación de las displasias pilosas
| Displasias pilosas con fragilidad |
| Moniletrix |
| Pseudomoniletrix |
| Pili torti |
| Síndrome de Menkes (kinky hair) |
| Síndrome de Netherton (tricorrexis invaginata) |
| Tricotiodistrofia |
| Tricorrexis nodosa |
| Cabello burbuja |
| Síndrome del cabello en anágeno suelto |
| Displasias pilosas sin o con poca fragilidad |
| Triconodosis |
| Pili annulati |
| Pseudopili annulati |
| Cabello lanoso difuso (woolly hair) |
| Cabello lanoso difuso parcial |
| Nevus de cabello lanoso (woolly hair nevus) |
| Ensortijamiento progresivo y adquirido del cabello |
| Pelo ensortijado parcial adquirido |
| Nevus de cabello erecto (straight hair nevus) |
| Pelo impeinable (pili canaliculi et trianguli) |
Modificado de Ferrando et al.2.
Se caracteriza por estrechamientos periódicos y regulares del tallo del pelo (fig. 1b). Clínicamente el cabello suele ser normal al nacimiento, pero a los pocos meses se vuelve corto, frágil y opaco, lo que provoca hipotricosis con predominio en la región occipital del cuero cabelludo, que está más expuesta a la fricción (fig. 3a). Los pacientes con moniletrix suelen tener una marcada hiperqueratosis folicular en el cuero cabelludo, que puede ser localizada y de predominio en la región occipital, o corresponder a un hallazgo generalizado. Esta hiperqueratosis folicular tan marcada también se puede observar en los pacientes con queratosis pilar decalvante (síndrome de Siemens)2. El moniletrix también puede afectar a las cejas (fig. 3b), las pestañas y el vello corporal. Se puede acompañar de alteraciones ungueales como coiloniquia7–10.
 Paciente con moniletrix. Se observa hipotricosis y pápulas con eritema perifolicular en el cuero cabelludo. B) Moniletrix en las cejas de la misma paciente. C) Imagen tricoscópica. D) Imagen al microscopio electrónico de un cabello con moniletrix.")
El moniletrix se transmite mediante herencia autosómica dominante (AD), con una alta penetrancia y expresividad variable. Se han descrito mutaciones en los genes KRT81, KRT83 y KRT86, que codifican para las queratinas Hb1, Hb3 y Hb6, lo que causaría una queratinización anómala de la corteza del tallo piloso7,8,11,12. También se ha descrito una forma de moniletrix con herencia autosómica recesiva (AR), causada por mutaciones en el gen DSG4, que codifica para la desmogleína 4, lo que provocaría alteraciones en la placoglobina y en los desmosomas de los tallos8. El moniletrix se ha asociado con queratosis pilaris, coiloniquia hereditaria y el síndrome de Holt-Oram7.
La exploración tricoscópica revela con claridad los estrechamientos del tallo piloso (fig. 3c), y además se puede observar desde eritema e hiperqueratosis perifolicular, hasta pápulas foliculares. Los tallos tienen un aspecto en «collar de cuentas» en donde se observan nidos ovalados que corresponden a áreas de diámetro normal, y estrechamientos regulares (fig. 3d), sin médula, por donde suelen ocurrir las fracturas7,13–16.
El pronóstico del moniletrix es variable. En la mayoría de los casos las alteraciones persisten durante toda la vida, aunque se han descrito remisiones parciales o totales asociadas al verano, al embarazo, al uso de anticonceptivos orales y a la edad avanzada17. En el tratamiento del moniletrix se han utilizado retinoides tópicos y orales, minoxidil tópico al 2% y al 5%, minoxidil oral y acetilcisteína, con resultados variables. En el caso de los retinoides orales se ha descrito la recurrencia del moniletrix al suspender el tratamiento18–20.
PseudomoniletrixClínicamente se presenta como áreas de alopecia difusa o limitada. Se caracteriza por cabellos cortos, arrosariados y que se rompen a los pocos milímetros de salir a la superficie2. Aparece a edades más avanzadas que el moniletrix. A diferencia del anterior, en el pseudomoniletrix no hay hiperqueratosis folicular y el tallo piloso presenta engrosamientos irregulares en lugar de estrechamientos, manteniendo zonas internodales de diámetro normal (fig. 1c). Al microscopio se observan engrosamientos indentados y aplanados21.
Camacho y cols. proponen la siguiente clasificación:
Pseudomoniletrix familiar de Bentley-Phillips (Tipo I)Es la forma hereditaria AD, y con penetrancia variable descrita en 1973 por Bentley-Phillips y Bayles22. Sólo afecta el cabello. Se inicia en la etapa prepuberal y puede dar origen a fracturas longitudinales.
Pseudomoniletrix adquirido en displasias pilosas (Tipo II)Presente en algunas displasias pilosas que asocian fragilidad del tallo, como por ejemplo moniletrix, pili torti, tricorrexis nodosa, pelo lanoso, cabello burbuja, ensortijamiento adquirido, acrodermatitis enteropática, etc.23. Clínicamente se puede manifestar como alopecia androgenética o tricotilomanía. El peinado y cepillado pueden agravar esta condición.
Pseudomoniletrix iatrogénico (Tipo III)Puede observarse en displasias pilosas con fragilidad capilar. También en cabellos finos y claros, por el traumatismo provocado al preparar el tricograma, por la compresión de las pinzas de Pean al realizar la tracción, o por la compresión durante el montaje en el portaobjetos24,25.
No requiere tratamiento ya que se trata de un artefacto de laboratorio21. El uso de gel de ecografía como líquido de inmersión puede dar un artefacto de falso aplanamiento de los tallos en la tricoscopia. Un efecto similar se produce por el uso de geles fijadores por parte de los pacientes, por lo que se recomienda no utilizar estos productos antes de realizar la tricoscopia14.
Pili tortiEste término se refiere a tallos aplanados y retorcidos sobre su propio eje con angulaciones de 90°, 180° y hasta 360°, con una frecuencia regular o irregular (fig. 1d)16. Se ha descrito que una vacuolización e irregularidades en el grosor de la vaina epitelial externa influirían en la vaina epitelial interna provocando los retorcimientos del pelo sobre su propio eje21. El pili torti puede afectar el cabello y a otras áreas pilosas del cuerpo como cejas, pestañas, vello axilar y púbico. Se produce un aumento en la fragilidad capilar, lo que lleva a la formación de fracturas e hipotricosis focal o difusa. Suele ocasionar fracturas longitudinales (tricoptilosis). A la exploración física se observa un cabello seco, corto, y la reflexión de la luz le proporciona un aspecto «en lentejuelas», debido a las angulaciones que permiten que solo algunos tramos del cabello reflejen la luz21.
El pili torti congénito se ha asociado a mutaciones en el gen BSC1L, ST14 y CDH3. Existen formas de pili torti con herencia AD y AR que se pueden manifestar de manera aislada, o asociada a otras enfermedades como los síndromes de Björnstad, Beare, Bazex, Menkes, Crandall, y la paquioniquia congénita tipo 22,17. Además, el pili torti, puede observarse como un defecto aislado en personas con pelo fino y rubio21. Algunos pacientes con pili torti mejoran después de la pubertad, pero en otros la alteración del tallo persiste durante toda la vida.
Los casos de pili torti adquirido son poco frecuentes y se han descrito en relación a déficits nutricionales, al uso de medicamentos como retinoides orales, erlotinib y a la enfermedad de injerto contra huésped26,27. Recientemente Ferrari y cols., describieron pili torti en la tricoscopia de una paciente con alopecia frontal fibrosante. Los autores proponen que el pili torti sería un signo de fibrosis y predictor de una menor respuesta a la infiltración de corticoides28.
Actualmente no existe un tratamiento eficaz para el pili torti y se recomienda evitar los traumatismos físicos o químicos del cabello.
Síndrome de Menkes (kinky hair)El síndrome de Menkes (SM) es una enfermedad neurodegenerativa, poco frecuente, provocada por alteraciones en el metabolismo del cobre29, descrita por el neurólogo pediátrico John Menkes en 196230. Se transmite mediante herencia recesiva ligada al X. La incidencia estimada en Europa es de 1 en 300.000 nacidos vivos29. El gen ATP7A, localizado en el cromosoma X (Xq21.1), 4,9 (9q31-q32), 14, y 18 (18.26.0cM), codifica una proteína transmembrana (ATP7A), que controla el transporte de cobre. La alteración en la absorción de cobre a nivel intestinal produce una disminución en los niveles de cobre y ceruloplasmina en el plasma y en órganos como el cerebro, el hígado, los huesos, el pelo y la piel6. Otras variantes patogénicas de esta mutación provocan manifestaciones más leves como el síndrome del cuerno occipital o una neuropatía motora29.
Típicamente los niños con SM se desarrollan de manera normal hasta los 2-3 meses de edad, y progresivamente presentan bajo peso, retraso psicomotor, hipotermia, hipotonía y convulsiones. Hallazgos típicos son las mejillas rechonchas, un marcado arco de cupido en el labio superior y cejas horizontales, lo que les da un aspecto de cara de «perdiz» (fig. 4a). Además tienen el cabello quebradizo, piel laxa, hiperlaxitud articular, hernias inguinales y divertículos vesicales y ureterales. Existen formas más leves de SM con escasas manifestaciones neurológicas, lo que puede retrasar el diagnóstico. También se han descrito casos de SM que debutaron con hematomas subdurales, defectos en la columna cervical, fracturas costales e irregularidades en las metáfisis de huesos largos, que inicialmente se confundieron con signos de maltrato infantil31.
 Paciente con síndrome de Menkes y kinky hair. B) Estudio con microscopía electrónica de los cabellos del mismo paciente. Se observan pili torti, angulaciones y tricorrexis nodosa.")
A la exploración tricoscópica se ven tallos de diámetro variable, con aplanamientos y giros irregulares (pili torti atípico), moniletrix, y/o tricorrexis nodosa2. Estos hallazgos se observan con mejor detalle al MO y al ME (fig. 4b).
El diagnóstico de SM se realiza mediante los hallazgos clínicos y la medición de los niveles de cobre y ceruloplasmina en suero29.
La evolución natural de la enfermedad es hacia una pérdida progresiva de las funciones neurológicas, lo que provoca la muerte durante los primeros años de vida29. El diagnóstico precoz es fundamental, ya que el tratamiento con cobre-histidina subcutáneo, podría mejorar el pronóstico a nivel neurológico2,29.
Síndrome de Netherton (Tricorrexis invaginata)El síndrome de Netherton (SN) es un trastorno congénito de la queratinización que se transmite mediante herencia AR y que afecta a la piel, el pelo y el sistema inmune32. Fue descrito por Comel (1949) y Netherton (1958). Se caracteriza por la triada de ictiosis (ictiosis lineal circunfleja u otros tipos de ictiosis congénita), displasias pilosas y signos de atopia (elevación de la IgE, hipereosinofilia, eccemas, rinitis, alergias alimentarias y asma)33,34. Son frecuentes las infecciones bacterianas recurrentes. El SN también se ha asociado con otros hallazgos como bajo peso, enteropatía, fallo renal y retraso intelectual34. La incidencia estimada del SN es de 1 en 100.000-200.000 nacidos vivos32. Mutaciones en el gen SPINK5 (cromosoma 5q32) provocan alteraciones en la proteína LEKTI, llevando a una sobreexpresión de tres tipos de calicreínas (KLK5, KLK7 y KLK14)6,34.
Clínicamente el cabello de los pacientes con SN es corto, sin brillo y quebradizo. Son característicos los tallos en «caña de bamboo» o tricorrexis invaginata, que corresponde a la invaginación del tallo sobre sí, y que se considera un hallazgo específico del SN (fig. 1e)35. En la tricoscopia, a bajo aumento, se observan múltiples engrosamientos a intervalos irregulares a lo largo del tallo piloso. A mayor aumento se evidencia la invaginación del segmento distal del tallo dentro de la porción proximal, formando una imagen en «bola dentro de una copa», que se considera patognomónica del SN. En el caso de la fractura del extremo distal, la porción proximal adopta un aspecto que se conoce como «golf tee» por la similitud con el soporte para las pelotas utilizadas en este deporte. Según Rudnicka y cols., es más fácil encontrar estos hallazgos tricoscópicos en las cejas de los pacientes con SN, por existir una densidad de tricorrexis invaginata 10 veces mayor que en el cuero cabelludo14,16. En el SN también es posible observar pili torti y tricorrexis nodosa, pero no son signos característicos y pueden mejorar con el tiempo e incluso resolverse por completo36.
Un estudio reciente de casos y controles que incluyó a ocho pacientes con SN, describió la presencia de un patrón en bandas al MO con luz polarizada que se observó con mayor frecuencia que la patognomónica tricorrexis invaginata33.
En la actualidad no existe un tratamiento específico para el SN, el pronóstico es pobre y la mortalidad es elevada durante los primeros años de vida debido a las pérdidas de agua, la deshidratación hipernatrémica y las infecciones bacterianas32,34. En los pacientes con SN los tratamientos están dirigidos a mejorar las manifestaciones cutáneas. Se han empleado los corticoides orales, retinoides, fototerapia, inhibidores de la calcineurina tópicos y lociones con lactato17. Las nuevas terapias dirigidas utilizadas en el tratamiento de la dermatitis atópica, como dupilumab, podrían tener utilidad en el SN aunque la evidencia aún es escasa para poder establecer recomendaciones al respecto34,37.
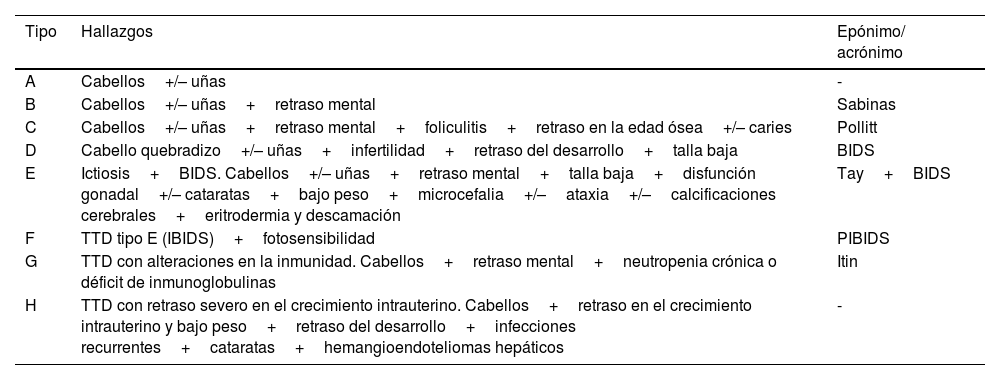
TricotiodistrofiaLa tricotiodistrofia (TTD) corresponde a un grupo heterogéneo de enfermedades (tabla 2)2. Los hallazgos más relevantes incluyen anormalidades en el cabello, microdolicocefalia, retraso psicomotor, ictiosis, signos de envejecimiento precoz y fotosensibilidad2,38. En los pacientes con TTD es frecuente observar alteraciones ungueales como onicodistrofia, coiloniquia, surcos, onicogrifosis, onicosquisis, coloración amarillenta y aumento en la curvatura longitudinal de las uñas (unguis inflexus)39. La TTD se transmite mediante herencia AR, y se han descrito cuatro genes implicados: XPD, XPB, p8/TTDA y TTDN1, los tres primeros con función en la transcripción, reparación y escisión de nucleótidos del ADN. A pesar de esto, los pacientes con TTD no tienen una mayor predisposición al cáncer a diferencia de los pacientes con xeroderma pigmentoso38–40. A la exploración clínica el cabello tiene un aspecto deslustrado, frágil, con hipotricosis y alopecia. Estos hallazgos también se encuentran en las cejas y pestañas. La TTD debe plantearse como un diagnóstico diferencial en los pacientes con alopecia congénita39.
Subtipos de tricotiodistrofia
| Tipo | Hallazgos | Epónimo/ acrónimo |
|---|---|---|
| A | Cabellos+/– uñas | - |
| B | Cabellos+/– uñas+retraso mental | Sabinas |
| C | Cabellos+/– uñas+retraso mental+foliculitis+retraso en la edad ósea+/– caries | Pollitt |
| D | Cabello quebradizo+/– uñas+infertilidad+retraso del desarrollo+talla baja | BIDS |
| E | Ictiosis+BIDS. Cabellos+/– uñas+retraso mental+talla baja+disfunción gonadal+/– cataratas+bajo peso+microcefalia+/–ataxia+/–calcificaciones cerebrales+eritrodermia y descamación | Tay+BIDS |
| F | TTD tipo E (IBIDS)+fotosensibilidad | PIBIDS |
| G | TTD con alteraciones en la inmunidad. Cabellos+retraso mental+neutropenia crónica o déficit de inmunoglobulinas | Itin |
| H | TTD con retraso severo en el crecimiento intrauterino. Cabellos+retraso en el crecimiento intrauterino y bajo peso+retraso del desarrollo+infecciones recurrentes+cataratas+hemangioendoteliomas hepáticos | - |
Modificado de Trichothiodystrophy: Update on the sulfur deficient brittle hair syndromes. Modificado de Itin et al.38,39.
Las alteraciones en el tallo piloso son obligatorias para establecer el diagnóstico de TTD. Para un adecuado diagnóstico se deben obtener muestras de diferentes zonas del cuero cabelludo y hacer una valoración con MO y ME39. Se observan fracturas transversales (tricosquisis), superficie y diámetros irregulares de los tallos, y hallazgos similares a tricorrexis nodosa y pili torti. El estudio mediante MO con luz polarizada revela la alternancia entre bandas claras y oscuras, que dan un aspecto en «cola de tigre» característico de la TTD (fig. 1f) (fig. 5)38. En los últimos años se han descrito hallazgos dermatoscópicos como los cabellos glomeruloides, que corresponden a pelos rotos a nivel del infundíbulo con ensortijamiento41. También la transiluminación polarizada utilizando dos dermatoscopios y un móvil, o un dermatoscopio y un espejo, permitirían ver la imagen de «cola de tigre» en la consulta42.

No existe un tratamiento específico para las alteraciones del cabello en los pacientes con TTD, aunque se han visto beneficios al evitar los traumatismos locales, utilizar minoxidil 2%, y emplear suplementos orales de cistina. La biotina oral no ha demostrado ser efectiva en estos casos39.
Tricorrexis nodosaLa tricorrexis nodosa (TN) corresponde a nódulos de fractura en el tallo piloso que clínicamente se caracteriza por un cabello fino y escaso. En la tricoscopia con líquido de inmersión se observan engrosamientos a lo largo del tallo piloso, que pueden acompañarse de fracturas. En la tricoscopia sin líquido de inmersión se observan pequeñas áreas blanquecinas que corresponden a las zonas de separación del tallo. A mayor aumento se observan numerosas fibras cortas en el nudo, como si se tratase de dos brochas que aproximan sus extremos entre sí (fig. 1g)8,14. La TN también se puede presentar como angulaciones abruptas del tallo piloso14. La exploración con MO o ME permite observar estos hallazgos con mayor detalle16.
Existen dos tipos de tricorrexis nodosa.
Tricorrexis nodosa distal: Es una alteración adquirida, que afecta a individuos de cabello largo, de manera difusa, y es causado por agentes físicos y químicos externos. Es más frecuente en mujeres jóvenes de cabello largo, y se asocia al cepillado excesivo y al uso de procedimientos estéticos. Clínicamente se reconocen pequeños engrosamientos blanquecinos en el extremo distal del pelo, que tienden a fracturarse de manera longitudinal, provocando lo que comúnmente se conoce como «puntas abiertas» o tricoptilosis2.
El tratamiento de la TN adquirida consiste en evitar el peinado excesivo, el calor, el viento, la sal o el uso de otros productos químicos, y preferir los champús suaves y el uso de acondicionadores43.
Tricorrexis nodosa proximal: Ocurre con menor frecuencia que la anterior. Los hallazgos clínicos son los mismos, pero se considera una patología más compleja, ya que pueden existir varios nudos en un mismo tallo y con frecuencia se asocia a hipotricosis y alopecia. Es más frecuente en personas de raza negra, que suelen mostrar hipotricosis en el cuero cabelludo y en otras localizaciones. La tricorrexis nodosa proximal también se ha descrito en relación al uso de fármacos como el trametinib y el inhibidor del factor de necrosis tumoral alfa44,45. La TN puede ser congénita y asociarse al síndrome de Menkes, tricotiodistrofia, moniletrix, arginosuccinuria congénita, citrulinemia, síndrome tricohepatoentérico, déficit de biotina e hipotiroidismo, entre otros3,6,7,16,46–48. En la TN congénita, heredada de manera AD, el cabello suele ser normal al nacimiento y durante los primeros meses de vida es reemplazado por un cabello frágil y escaso17.
Cabello burbujaEs una displasia pilosa adquirida que se observa con mayor frecuencia en mujeres. La paciente habitualmente consulta por una placa de cabello corto y frágil3. En la exploración con tricoscopia, MO y ME, se observan cavidades de aire o vacuolas en la corteza del tallo piloso que se producen por el paso de agua a altas temperaturas en el interior del tallo, lo que provoca la hidrólisis de la queratina y la expansión local del aire (fig. 1h). Se asocia al uso de secadores de pelo, rizadores y planchas de pelo a temperaturas superiores a 125°C43. El cabello burbuja se puede asociar a tricorrexis nodosa y tricoptilosis. Los hallazgos mejoran al reducir el uso de químicos y de calor sobre el cabello43,49.
Síndrome del cabello en anágeno sueltoSe produce por una pérdida de anclaje entre el tallo piloso y el cuero cabelludo2. Es más frecuente en niñas rubias entre los 3 y 6 años (fig. 6a), aunque se ha descrito en pacientes de todos los fototipos y también en la edad adulta. Se asocia a mutaciones en SHOC2 (10q.25) y KRT75.77,78 (12q.13)6.
 Hipotricosis en una niña con cabello anágeno suelto. B) Raíces anagénicas torsionadas y «ruffling» de la vaina interna del folículo piloso.")
Típicamente el cabello se desprende con facilidad frente a una tracción suave, sin causar dolor, pudiendo provocar una alopecia difusa. Los padres comentan que no es necesario cortarles el pelo, porque crece muy poco50.
El síndrome del cabello en anágeno suelto se ha clasificado en tres tipos según su fenotipo51:
Tipo A: caracterizado por una disminución en la densidad capilar.
Tipo B: caracterizado por un cabello difícil de peinar.
Tipo C: cabellos de apariencia normal, pero con un aumento en la caída.
En el MO se observan bulbos distróficos en anágeno o «en palo de golf», sin vainas, y con una cutícula enrollada a nivel proximal (rufling) (fig. 1i) (fig. 6b)52.
El síndrome del cabello en anágeno suelto no requiere tratamiento, ya que mejora espontáneamente con los años. Se ha descrito buena respuesta al minoxidil tópico al 2% y 5%2,53.
A la Dra. Ana Martín-Santiago por sus comentarios durante la redacción del manuscrito y la colaboración con imágenes clínicas. Al Dr. Carlos Saus por su dedicación y las imágenes microscópicas. A Marina Cascales por su gentileza en la elaboración de las representaciones gráficas.


