

Con el desarrollo de la enfermedad molecular, el diagnóstico y la comprensión de los tumores melanocíticos ha experimentado un avance descomunal en los últimos años. Esto ha significado la aparición de conceptos de difícil asimilación en el mundo clínico, el cual no está siempre en contacto directo con las técnicas genéticas de laboratorio. Al mismo tiempo, sin embargo, al clínico se le está exigiendo una terapéutica basada en muchas ocasiones en los hallazgos moleculares más recientes de un tumor melanocítico. El presente artículo explora los conceptos moleculares más recientes de la clasificación en rutas patogénicas de los tumores melanocíticos, incluidas las formas intermedias conocidas como melanocitomas, y repasa las técnicas auxiliares usadas en el estudio de estos tumores, discutiendo los resultados más complejos, sus limitaciones, y sus solapamientos.
The advent of molecular pathology has fueled unprecedented advances in the diagnosis and understanding of melanocytic tumors. These advances, however, have also generated concepts that may be difficult to grasp for clinical practitioners, who are not always conversant with the array of genetic techniques employed in the laboratory. These same practitioners, however, are being increasingly called on to provide treatments that are often based on the latest molecular findings for melanocytic tumors. We review the most recent concepts in the pathway classification of melanocytic tumors, including intermediate lesions known as melanocytomas. We examine the genetic and molecular techniques used to study these tumors, look at where they overlap, and discuss their limitations and some of the most difficult-to-interpret results.
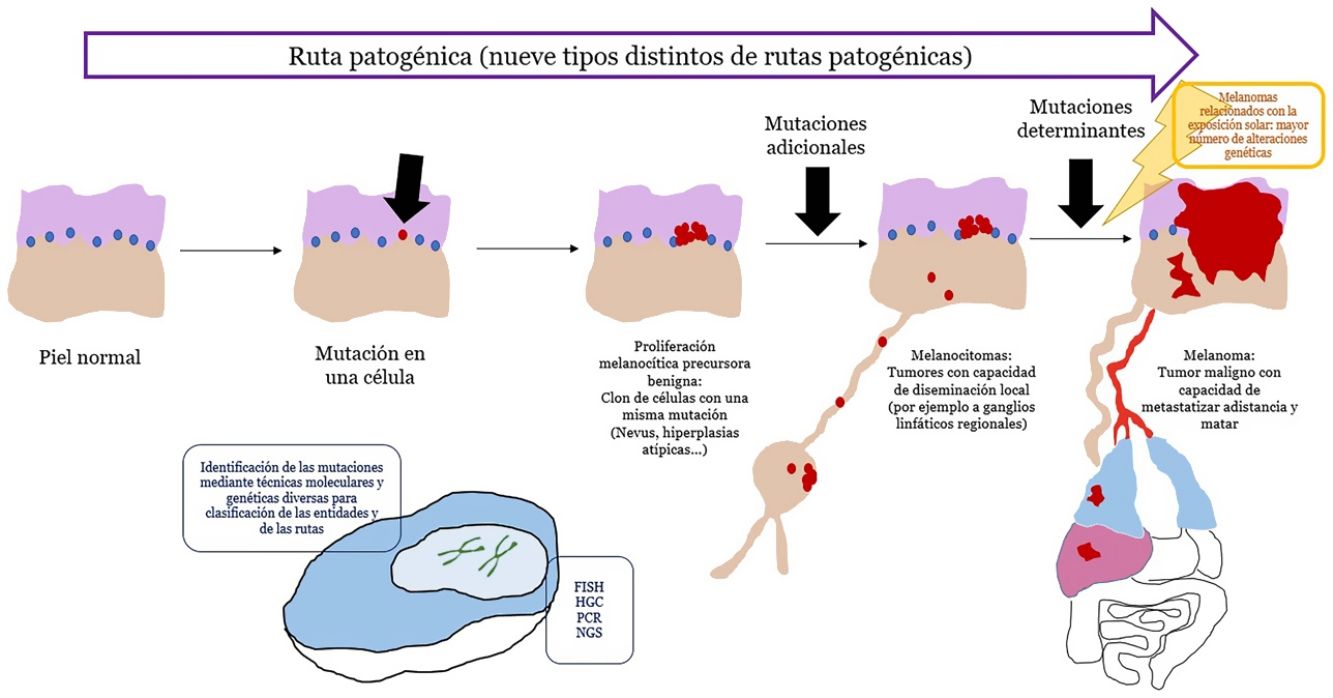
En las últimas décadas, nuestro conocimiento etiopatogénico del melanoma se ha incrementado considerablemente gracias a técnicas genéticas y moleculares. Esto ha permitido descifrar un puzle complejo en el que la dualidad nevus benigno/melanoma maligno como 2únicas opciones, ha dado paso a un espectro con varios pasos intermedios.
También nos ha llevado a reconocer que el melanoma no es uno, sino que existen varios tipos de melanoma que pueden desarrollarse a través de rutas patogénicas diferentes y sin relación entre sí.
El reconocimiento cada vez más exacto de las formas intermedias entre nevus y melanoma nos ha llevado a diagnósticos de dificultad conceptual de entidades que no son benignas o malignas en términos absolutos, sino una progresión intermedia hacia el melanoma. Esto último se comprendió mejor con el desarrollo del ganglio centinela y la evidencia de tumores melanocíticos que se diseminan localmente, por ejemplo, a ganglios linfáticos, sin capacidad para matar al individuo.
Este panorama conceptual complejo ha necesitado recuperar para su arsenal descriptivo, términos que se consideraban casi extintos en la literatura, como el de melanocitoma. En consecuencia, existe una demanda del dermatólogo por la comprensión del estado histopatológico de tan actual y dinámico mapa.
El presente artículo tiene como objetivo la explicación y actualización de algunos de estos conceptos desde el punto de vista del patólogo, adaptados a la aplicación clínica del dermatólogo.
Los tumores melanocíticos son resultados de alteraciones genéticasComo tantos otros tumores, los melanocíticos, bien sean benignos o malignos, son el resultado de alteraciones genéticas de diversos tipos.
Los nevus suelen ser el resultado de una única mutación en un melanocito, que da lugar a una expansión clonal benigna. Los nevus adquiridos aparecen por mutaciones de melanocitos que normalmente ya han alcanzado la epidermis, dando lugar a nevus junturales primero, que pasarán a ser compuestos y luego intradérmicos. La gran mayoría de nevus adquiridos presentan una mutación del codón 600 del gen BRAF1,2.
Por el contrario, en los nevus congénitos, la mutación se produce en algún momento durante la migración del melanocito hasta la epidermis desde la cresta neural. Cuanto más cerca esté el melanocito de la epidermis, menor será el tamaño del nevus, mientras que si la mutación se produce en un momento precoz de la migración, el nevus congénito puede ser muy extenso y afectar a múltiples zonas corporales. Al contrario que los nevus adquiridos, los nevus congénitos suelen mostrar mutaciones en el gen NRAS, frecuentemente en el codón 613,4.
Uno de los factores más importantes —pero no el único— en la tumorigénesis del melanoma es la exposición solar5-7. Aquellos melanomas desarrollados en el contexto de una intensa y prolongada exposición solar son los que muestran más aberraciones genéticas8-13. Por el contrario, aquellos melanomas probablemente poco o nada relacionados con la exposición solar —como el melanoma acral— muestran una carga mutacional baja13-15. Los rayos UVA son menos carcinógenos que los UVB: mientras que los primeros inducen la oxidación de la guanina en el ADN, los segundos generan dímeros oncogénicos de ciclobuteno16,17.
Si algo hemos aprendido durante todas estas décadas de examen de melanomas al microscopio óptico es la amplia variedad morfológica que estos tumores presentan: melanomas de extensión superficial, nodular, spitzoides, acrales, etc- Sin embargo, y a pesar de estas variaciones, los melanomas suelen tener una patogenia genómica limitada, que afecta a un grupo reducido de vías moleculares. Las principales vías son18-23:
- 1.
La vía MAPK/ERK (cinasas MAP, también conocidas como ERK), que comprende varias cinasas reguladas por factores extracelulares. Entre las MAPK tenemos MAP2K1, MAP3K, MAP2K o TRK. En esta ruta, la señal es transmitida hasta el núcleo a través de proteínas como RAS o RAF y factores de transcripción como MYC.
- 2.
La vía PI3K/AKT/mTOR de la fosfatidilinositol-3-cinasa.
- 3.
La vía del oncogén p53.
- 4.
La vía del MITF.
- 5.
La vía del NFkB.
- 6.
La vía del ciclo celular (Punto G1/S).
- 7.
La vía del WNT y la regulación apoptótica.
- 8.
La vía de la remodelación de cromatina SWI/SNF.
Desde un punto de vista genético, lo que distingue a unos tumores melanocíticos de otros son los mecanismos de alteración de estas vías. Por ejemplo, la vía MAPK puede estar alterada por mutaciones en BRAF, NRAS o NF124-26 (frecuentes en nevus comunes y en melanoma asociado a baja exposición solar), mientras que la misma vía MAPK puede alterarse por mutaciones en HRAS o por fusiones en los genes de las cinasas en el caso de los tumores de Spitz27.
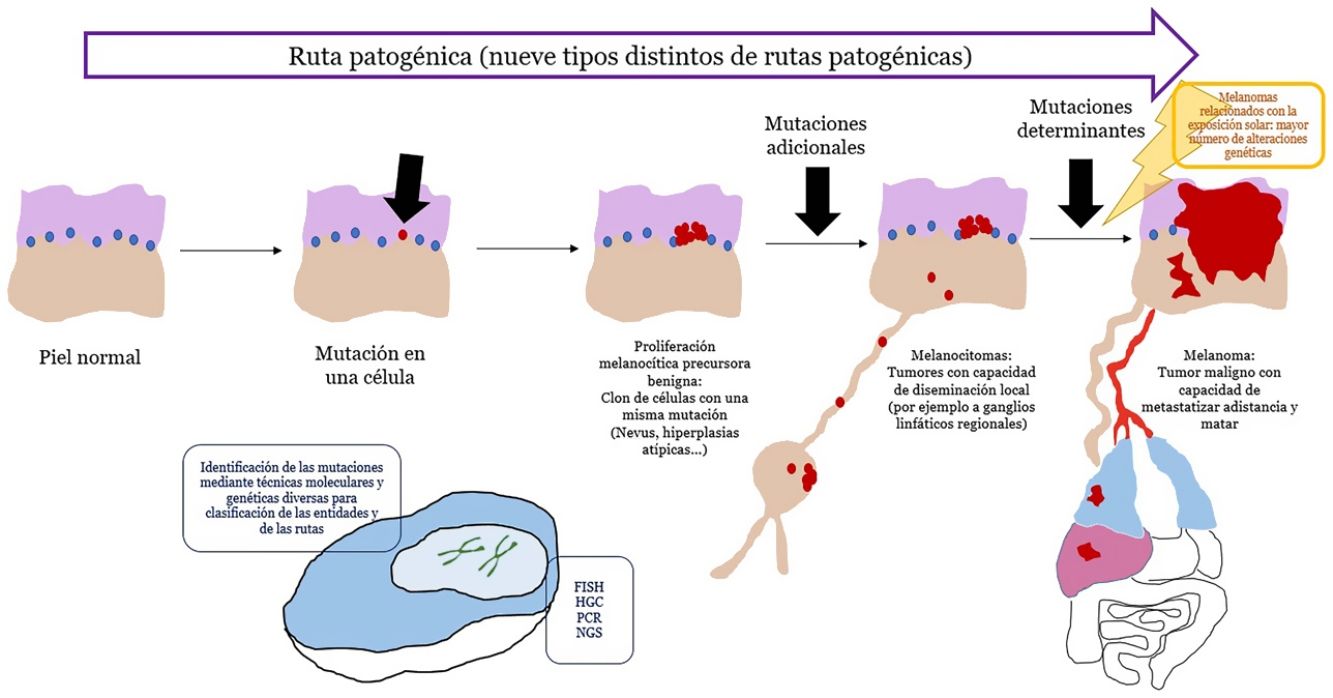
El concepto de rutas patogénicasA pesar de que los nevus son tumores con mutaciones, estas son insuficientes para transformar a la célula en invasiva o metastásica. Por el contrario, los nevus melanocíticos tienen un crecimiento limitado debido al proceso de senescencia, mediante el cual una célula fracasa en su reingreso en el ciclo celular, no perpetuando, por lo tanto, el proceso mitótico28. Varios mecanismos se encargan de inducir senescencia, evitando el desarrollo de tumores incontrolables. De entre los mecanismos más conocidos está el representado por los genes supresores de tumores p53 y p1629.
Si un nevus experimenta cambios genéticos y epigenéticos adicionales, puede escapar a los mecanismos de senescencia, convirtiéndose en una lesión intermedia con bajo o alto riesgo de progresar o, finalmente, en un melanoma. Esta interpretación de la naturaleza de los tumores melanocíticos ha derivado en la identificación de varias rutas patogénicas30-32. Cada ruta tendría una contrapartida benigna névica o bien un precursor hiperplásico benigno, así como formas intermedias de malignidad hasta llegar al melanoma. Algunas de las rutas están relacionadas con la exposición solar como causa etiológica, pero otras no33. Este modelo tiene una gran carga hipotética, ya que los precursores benignos de alguna de las entidades todavía no están bien categorizados.
Las rutas no siempre se inician en el primer paso. Por ejemplo, un melanocitoma (a pesar de considerarse un paso intermedio entre nevus y melanoma) puede surgir de novo. De hecho, durante años, el axioma general ha sido que la mayoría de los melanomas se originan de novo, mientras que solo una minoría se originan a partir de nevus34,35. Este axioma requiere algunas puntualizaciones, ya que el melanoma más frecuente en nuestro medio, el melanoma lentigo maligno, se origina a partir de pasos mutacionales previos in situ (aunque no sean «névicos»), como el lentigo maligno o similares. El surgimiento de novo de un melanoma que posea directamente propiedades infiltrantes es difícil de explicar genéticamente, ya que requeriría un cúmulo de eventos genéticos catastróficamente ocurridos de golpe, como la cromoptisis o la cromoplexia36, ambas infrecuentes. Alternativamente, algunos autores han sugerido que melanocitos histológicamente normales, podrían, sin embargo, tener mutaciones iniciales que no induzcan proliferación (p. ej., en CDKN2A o en el promotor de TERT)36,37. Además, hay autores que señalan que nuestro conocimiento de este proceso puede estar sesgado: la ausencia de un nevus remanente en una extirpación de un melanoma no necesariamente significa que dicho melanoma no se haya originado en un nevus: el crecimiento del melanoma o bien la respuesta inmunitaria a este pueden haber destruido los restos névicos iniciales36.
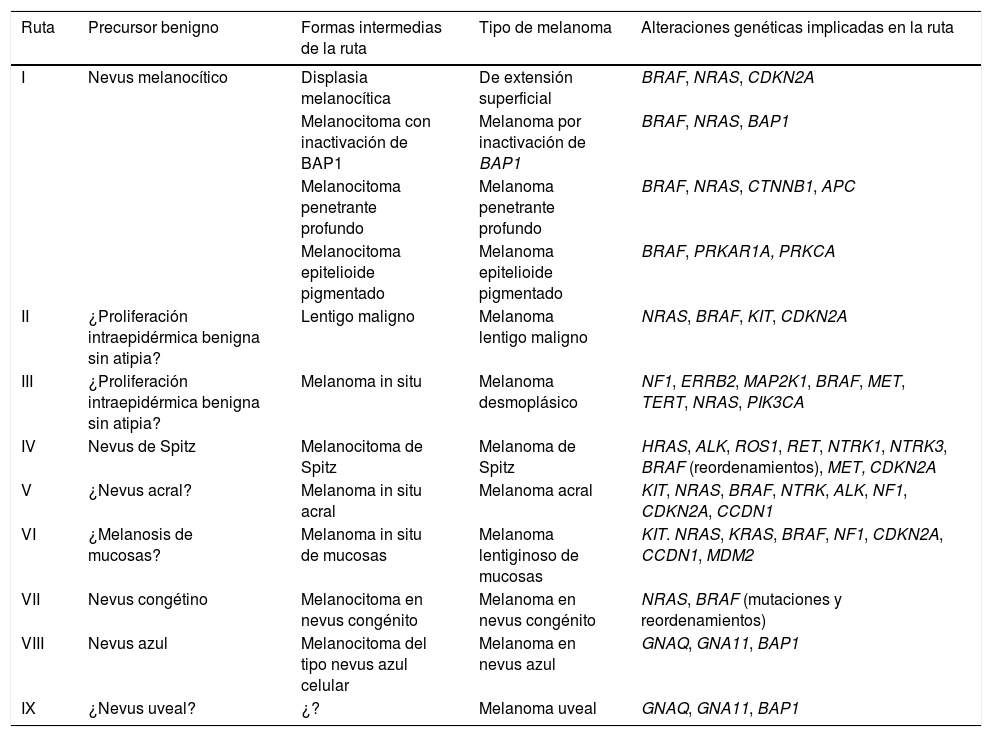
Las rutas reconocidas por la OMS se detallan a continuación33,38 (tabla 1).
Rutas patogénicas simplificadas identificadas por la OMS en la formación de los distintos tipos de melanoma
| Ruta | Precursor benigno | Formas intermedias de la ruta | Tipo de melanoma | Alteraciones genéticas implicadas en la ruta |
|---|---|---|---|---|
| I | Nevus melanocítico | Displasia melanocítica | De extensión superficial | BRAF, NRAS, CDKN2A |
| Melanocitoma con inactivación de BAP1 | Melanoma por inactivación de BAP1 | BRAF, NRAS, BAP1 | ||
| Melanocitoma penetrante profundo | Melanoma penetrante profundo | BRAF, NRAS, CTNNB1, APC | ||
| Melanocitoma epitelioide pigmentado | Melanoma epitelioide pigmentado | BRAF, PRKAR1A, PRKCA | ||
| II | ¿Proliferación intraepidérmica benigna sin atipia? | Lentigo maligno | Melanoma lentigo maligno | NRAS, BRAF, KIT, CDKN2A |
| III | ¿Proliferación intraepidérmica benigna sin atipia? | Melanoma in situ | Melanoma desmoplásico | NF1, ERRB2, MAP2K1, BRAF, MET, TERT, NRAS, PIK3CA |
| IV | Nevus de Spitz | Melanocitoma de Spitz | Melanoma de Spitz | HRAS, ALK, ROS1, RET, NTRK1, NTRK3, BRAF (reordenamientos), MET, CDKN2A |
| V | ¿Nevus acral? | Melanoma in situ acral | Melanoma acral | KIT, NRAS, BRAF, NTRK, ALK, NF1, CDKN2A, CCDN1 |
| VI | ¿Melanosis de mucosas? | Melanoma in situ de mucosas | Melanoma lentiginoso de mucosas | KIT. NRAS, KRAS, BRAF, NF1, CDKN2A, CCDN1, MDM2 |
| VII | Nevus congétino | Melanocitoma en nevus congénito | Melanoma en nevus congénito | NRAS, BRAF (mutaciones y reordenamientos) |
| VIII | Nevus azul | Melanocitoma del tipo nevus azul celular | Melanoma en nevus azul | GNAQ, GNA11, BAP1 |
| IX | ¿Nevus uveal? | ¿? | Melanoma uveal | GNAQ, GNA11, BAP1 |
Ruta II (baja exposición solar): acaba en el melanoma tradicionalmente conocido como «de extensión superficial». El 90% de los tumores melanocíticos de esta ruta presentan mutaciones en el gen BRAF, siendo la más frecuente la mutación V600E. Las mutaciones en NRAS son mucho menos frecuentes.
A este tipo de melanoma se puede llegar por distintos caminos:
- –
A través de una displasia de bajo grado primero y de alto grado después.
- –
A través de inactivación del gen BAP1 primero y de mutaciones adicionales después.
- –
A través de un nevus penetrante profundo primero, con mutaciones adicionales después.
- –
A través de un melanocitoma epitelioide pigmentado primero, con mutaciones adicionales después.
En esta ruta no es infrecuente que en las fases próximas a la malignidad haya pérdida de expresión de p16 por alteraciones del gen CDKN2A39. Sin embargo, la evaluación de p16 requiere algunas consideraciones. Aunque p16 se considera un marcador de senescencia y, por lo tanto, tranquilizador cuando se expresa40-42, no está, sin embargo, expresado, en melanocitos normales. Su expresión puede inducirse frente al estrés celular43. Puede haber nevus comunes que expresen poco p16, sin que eso signifique una inactivación de CDKN2A.
Ruta II (alta exposición solar): acaba en el melanoma de tipo lentigo maligno, iniciándose en el lentigo maligno o en otros tipos de melanoma in situ.
Ruta III (también de alta exposición solar): acaba en el melanoma desmoplásico, iniciándose como melanoma in situ.
Ruta IV: acaba en el melanoma de Spitz, iniciándose en el nevus de Spitz benigno44,45. Suele tener mutaciones activadoras de HRAS (con o sin amplificaciones concomitantes del 11p, que es donde está localizado el gen HRAS), reordenamientos de los genes del receptor de las tirosin-cinasas (ROS1, ALK, MET, NTRK1, NTRK3 o RET) o reordenamientos de los genes de las serin/treonin-cinasas involucradas en la ruta MAPK (BRAF, RAFF1, MAP3K8)37,46. Los reordenamientos en las cinasas son mutuamente excluyentes (la presencia de uno excluye los otros). Los reordenamientos de BRAF no implican mutaciones en el gen (que son, por el contrario, frecuentes en la ruta I). En ocasiones, en los genes adyacentes a las zonas reordenadas de los tumores de Spitz, incluso benignos, pueden producirse cambios numéricos (p. ej., amplificaciones), sin necesidad de que impliquen mayor riesgo de progresión.
Una característica curiosa de la ruta de los tumores de Spitz, es que pueden presentar inactivación del gen CDKN2A (en muchos casos como única alteración genética), con pérdida de la expresión de p16, un hecho que sería preocupante en tumores melanocíticos de cualquier otra ruta47. Se necesitan, por lo tanto, mutaciones adicionales a la inactivación de CDKN2A para la transformación maligna. Por ejemplo, alteraciones en el promotor del TERT.
Otra característica común de este grupo es que las lesiones malignas pueden conservar las mutaciones previas de las lesiones benignas precursoras. Por lo tanto, aunque las mutaciones HRAS son típicas de los nevus de Spitz, demostrarlas no es garantía de benignidad: un melanoma de Spitz puede también tenerlas48,49. Sin embargo, puesto que las mutaciones en HRAS están presentes en menos de un 1% de melanomas37 su evidencia resulta tranquilizadora en un tumor de morfología Spitz.
Ha habido intentos de correlacionar las alteraciones genéticas evidenciadas en los tumores de Spitz, con los hallazgos histopatológicos en la hematoxilina-eosina50, empezando por la sugerencia de que las mutaciones HRAS activan con más vigor la vía PI3K/AKT/mTOR que las mutaciones BRAF o NRAS y, por lo tanto, inducen células tumorales de mayor tamaño y menos pigmentación que las vistas en los nevus comunes o congénitos37. Aunque esas correlaciones no son siempre fáciles de evaluar, se sabe, por ejemplo, que las mutaciones en HRAS están asociadas con caracteres desmoplásicos; los reordenamientos en MAP3K8, con predominio de células epitelioides, células multinucleadas y ulceración; los reordenamientos en RET con una silueta tumoral en placa y con nidos celulares poco cohesivos; los reordenamientos en NTRK3, con tumores pigmentados; los reordenamientos en NTRK1 con presencia de seudorrosetas; los reordenamientos en ROS1, con abundancia de cuerpos de Kamino, y los reordenamientos en ALK, con tumores grandes polipoides o cupuliformes, muchas veces con un hábito arquitectural fasciculado y en ocasiones con prominente depósito mixoide. Sin embargo, estas correlaciones no son completas. Así, por ejemplo, un fenotipo desmoplásico puede encontrarse sin asociación a mutación de HRAS o amplificación 11p y, por el contrario, asociarse a fusiones de cinasas como ROS1.
Los reordenamientos en BRAF, al contrario que las fusiones en otras cinasas, parecen ser eventos bastante terminales en la ruta, es decir, asociados más a la parte maligna del espectro, ya que se han visto preferentemente en melanomas de Spitz y en melanocitomas de Spitz y, en mucha menor medida, en nevus de Spitz.
Ruta V: acaba en el melanoma acral, iniciándose quizá en nevus acrales o en lesiones precursoras acrales malignas in situ.
Ruta VI: se inicia en la melanosis con displasia y acaba en el melanoma lentiginoso de mucosas. No sabemos si la melanosis no displásica podría ser el inicio de la ruta.
Ruta VII: esta ruta se refiere a los melanomas originados sobre nevus congénito. Sobre estos últimos pueden aparecer nódulos de crecimiento, debidos a mutaciones adicionales a las del nevus, por lo que se consideran melanocitomas de riesgo bajo/intermedio de evolución a melanoma. La ruta acaba en el melanoma sobre nevus congénito. En esta ruta, las lesiones melanocíticas contienen mutaciones hotspot NRAS, y menos frecuentemente, mutaciones en BRAF.
Ruta VIII: se inicia con el nevus azul, pasando por el nevus azul celular (incluido dentro de los melanocitomas de riesgo bajo/intermedio de evolución a melanoma), el nevus azul celular atípico y, finalmente, el melanoma sobre nevus azul, una entidad rara. Esta ruta se debe fundamentalmente a oncogenes que activan la vía de la G-alfa-q, siendo la alteración genética más común, las mutaciones activadoras en GNAQ o en GNA11. En las últimas fases de malignización puede mostrar mutaciones en el gen supresor de tumores BAP1.
Ruta IX: esta es la ruta del melanoma uveal. Se desconoce cuáles puedan ser los pasos histológicos previos hasta su transformación en melanoma. Puede presentar mutación del gen supresor de tumores BAP1.
Este esquema de las rutas patogénicas, permite fácilmente explicar por qué el número de nevus es mayor en individuos con piel clara, o con síndromes de susceptibilidad al melanoma, puesto que son más predispuestos a una mutación en sus melanocitos. También por qué episodios de exposición intermitente al sol durante la infancia están asociados con un mayor número de nevus. Y también por qué un mayor número de nevus implica un mayor riesgo de melanoma.
Se debe destacar que la OMS considera que no todos los tumores melanocíticos muestran el mismo riesgo de progresión a melanoma. Algunos son catalogados como de riesgo bajo/intermedio (como las melanosis atípicas, los melanocitomas en nevus congénitos, el nevus penetrante profundo o el melanocitoma de tipo nevus azul), mientras otros están considerados de riesgo intermedio/alto (como el melanocitoma por inactivación de BAP1, el melanocitoma penetrante profundo, el melanocitoma epitelioide pigmentado, el lentigo maligno, el melanoma in situ —bien sea acral, de mucosas o en nevus congénito— o el nevus azul celular atípico). También explica por qué algunas lesiones aparentemente poco aparatosas desde el punto de vista histopatológico (como, por ejemplo, un melanoma acral in situ, a veces con cambios histopatológicos muy sutiles, difíciles de diferenciar de un nevus), progresan rápidamente hasta melanoma invasivo si no se diagnostican correctamente a tiempo.
El concepto de melanocitomaEl término melanocitoma se ha consolidado en la clasificación de la OMS para referirse a tumores melanocíticos con una posición intermedia en la evolución de una lesión a lo largo de una ruta patogénica desde la benignidad hacia la malignidad51. Se les ha segregado como grupo individualizado distinto al de los nevus, al comprobar que presentan mutaciones driver múltiples. En ocasiones puede verse un melanocitoma coexistiendo en el seno de un nevus melanocítico común; señal de que dentro del nevus ha surgido una nueva población clonal del melanocitoma, como resultado de mutaciones o reordenamientos adicionales. Sin embargo, a veces los melanocitomas surgen de novo, por lo que el paso mutacional previo de los nevus comunes, no siempre es necesario.
Muchos de los melanocitomas tienen capacidad de diseminación a los ganglios linfáticos regionales, sin metastatizar más allá. La OMS los divide en 2grupos de riesgo de progresión a melanoma: los de grado bajo/intermedio y los de grado intermedio/alto.
En la actualidad, estos son los principales tipos de melanocitoma reconocidos:
- -
Melanocitoma por inactivación del BAP152: esta lesión melanocítica está incluida en la ruta patogénica del melanoma por exposición solar intermitente (ruta I). Los tumores de este grupo muestran inactivación bialélica del gen supresor de tumores BAP1, la mayoría de las veces por asociación de una mutación truncante en uno de los alelos del cromosoma 3 (locus 3p21) y pérdida completa o parcial del otro. Sería un paso intermedio entre el nevus de Wiesner (también llamado BAPoma) y los melanomas con inactivación de BAP1. El concepto de melanocitoma por inactivación de BAP1 es fundamentalmente genético y los hallazgos histológicos por sí solos no siempre permiten distinguir con firmeza un BAPoma de un melanocitoma. Por ello, algunos autores han sugerido gradar la atipia como información adicional en este tipo de diagnósticos52. De igual manera, el melanoma por inactivación de BAP1, suele presentar alteraciones histopatológicas suficientes que permitan sugerir ese diagnóstico. Pero ante cualquier tumor con inactivación de BAP1 y cierto grado de atipia, la demostración de la ausencia de alteraciones genéticas típicas de un melanoma permitirá descartar dicha progresión. La inactivación de BAP1 puede coexistir con mutaciones de NRAS y BRAF. Por lo tanto, y en contra de lo que se pensaba en el pasado, no se trata de una variante de tumor de Spitz. En la mayoría de los casos se puede demostrar la inactivación del gen mediante inmunohistoquímica, observando la pérdida de tinción nuclear53. Sin embargo, la inmunohistoquímica no detecta aquellos casos con mutaciones «sin sentido» en donde se sigue produciendo una proteína no funcional.Algunos individuos presentan una mutación germinal de BAP1 en uno de los alelos, manifestando un síndrome de predisposición tumoral multiorgánico54, con un mayor riesgo de presentar mesoteliomas, carcinomas renales, colangiocarcinomas, carcinomas basocelulares y, frecuentemente, melanomas cutáneos y uveales.La OMS considera que el riesgo de transformación de un melanocitoma con mutación BAP1 a un melanoma es medio-alto, aunque esto no concuerda con la experiencia de todos los autores37. Este tipo de melanocitoma generalmente no tiene mitosis o muestra ocasionales mitosis. Un índice alto de mitosis es un signo alto de sospecha de que la transformación a malignidad ya ha ocurrido.Se debe recordar que las mutaciones en BAP1 no son exclusivas de este melanocitoma. Pueden también verse en otros tipos de tumores melanocíticos, por lo que una inmunohistoquímica negativa para BAP1 no garantiza el diagnóstico de este melanocitoma. Es más, las mutaciones en BAP1 son características de las fases tardías (de malignización) de otras rutas patogénicas (como el melanoma uveal55 o el melanoma sobre nevus azul56), por lo que tendrán que considerarse con cautela y preocupación en esos contextos.
- -
Melanocitoma penetrante profundo57: también perteneciente a la ruta patogénica I, sería una lesión intermedia entre el nevus penetrante profundo y el melanoma desarrollado en nevus penetrante profundo. Desde un punto de vista histológico, el melanocitoma muestra algunos rasgos atípicos que el nevus no, tales como gran tamaño, asimetría, disposición «en sábana» de melanocitos, mitosis y atipia citológica severa. Sin embargo, un melanocitoma no mostrará todavía las alteraciones genéticas propias de un melanoma, por lo que en los casos de melanocitomas más atípicos, la ausencia de progresión a melanoma deberá ser demostrada mediante técnicas especiales (véase más adelante). Este grupo muestra mutaciones (generalmente puntuales) que inactivan la betacatenina, lo cual puede demostrarse mediante positividad inmunohistoquímica para el marcador de betacatenina58 o, alternativamente, para LEF1 (el receptor nuclear de betacatenina)59. En el caso de nevus comunes, la positividad para betacatenina se observa solo en los melanocitos próximos a la epidermis y a los anejos, mientras que en el melanocitoma penetrante profundo la betacatenina tiñe de modo uniforme y difuso los citoplasmas de los melanocitos tumorales y a veces también intensamente los núcleos. La betacatenina no es el único mecanismo por el que se genera un melanocitoma penetrante profundo. Otro mecanismo alternativo es, por ejemplo, la pérdida bialélica de función del gen APC (adenomatous polyposis coli)57. La OMS considera que el riesgo de transformación de un melanocitoma penetrante profundo a un melanoma es medio/alto, como queda demostrado con casos que, tras progresión, causaron la muerte del paciente60.
- -
Melanocitoma epitelioide pigmentado: también es un melanocitoma de ruta patogénica I y suele tener mutaciones en BRAF. Se trata de un melanocitoma indolente que, a pesar de mostrar invasiones ganglionares en un número elevado de pacientes, no suelen producir metástasis a distancia, sin que se hayan informado muertes debidas a este tumor. A pesar de su parecido histológico con los nevus azules, no está genéticamente relacionado con ellos.Dentro de este grupo hay una variante conocida como melanocitoma con inactivación del gen supresor de tumores PRKAR1A61,62 (mal llamado en el pasado nevus azul epitelioide), relacionado con el complejo de Carney63. La inactivación de PRKAR1A puede demostrarse mediante la negatividad para el anticuerpo anti-PRKAR1A en estudio inmunohistoquímico. Paralelamente, otros melanocitomas epitelioides pigmentados muestran combinación de mutaciones en BRAF con inactivación de PRKAR1A. Por último, otros tumores de este grupo presentan otras alteraciones genéticas como mutaciones en GNAQ, MAP2K1 o incluso fusiones en PRKCA. Está por definir si tales tumores pueden seguir incluyéndose en este grupo de melanocitomas indolentes o si representan otro tipo de tumores melanocíticos pertenecientes a otras rutas patogénicas.La OMS considera que el riesgo de transformación de los melanocitomas epitelioides pigmentados a un melanoma es medio/alto.
- -
Melanocitoma de Spitz45: pertenece a la ruta patogénica IV que va desde los nevus de Spitz hasta el melanoma de Spitz. El melanocitoma de Spitz presenta alteraciones del gen CDKN2A que codifica para p16 y p1444. Por ello, mientras que la pérdida de expresión de p16 es un evento preocupante en muchos tumores melanocíticos, sobre todo de ruta patogénica I, es bastante común en la ruta patogénica de los Spitz. La inactivación genética de CDKN2A sucede a menudo por deleción homocigota, pero también puede suceder por mutación truncadora en un alelo, seguida de pérdida de heterocigosidad, a menudo debido a pérdida de parte del cromosoma 9. Puesto que puede haber tumores con retención de una copia de CDKN2A que permanece activa, la FISH no nos proporciona información de tales casos, debiendo complementarse con la inmunohistoquímica para p16 (que se mantendrá positiva). Esta última puede ser citoplásmica o nuclear y, a menudo, se ve en «mosaico», es decir, células positivas y negativas de modo alternante. Los melanocitomas de Spitz tienen una alta tendencia a invadir ganglios linfáticos regionales, pero con un comportamiento global benigno y baja tasa de recurrencia. La OMS considera que su riesgo de transformación a un melanoma es bajo/medio. Mientras que el melanocitoma de Spitz muestra una deleción heterocigota de CDKN2A, una deleción homocigota debería ser altamente sospechosa de melanoma de Spitz. También mutaciones adicionales, como en el promotor del TERT, pueden alertar sobre un progreso a la malignidad en un melanocitoma de Spitz.
- -
Melanocitoma en nevus congénito64: se trata de una lesión nodular de carácter intermedio aparecida sobre un nevus congénito, por lo tanto, perteneciente a la ruta VII. En la clasificación de la OMS, este melanocitoma es sinónimo del término «nódulo en nevus congénito». A pesar de su aspecto histológico alarmante, estos melanocitomas suelen mostrar pérdidas o ganancias de cromosomas enteros, al contrario que los melanomas, que muestran alteraciones numéricas de segmentos cromosómicos65.La OMS considera que su riesgo de transformación a un melanoma es bajo/medio.
- -
Melanocitoma azul: también llamado, en el pasado, nevus azul celular66. Como tumor perteneciente a la ruta VIII, suele mostrar mutaciones que activan la vía G-alfa-q (mutaciones en GNAQ). Al contrario que el melanocitoma penetrante profundo, que muestra expresión nuclear de betacatenina, el melanocitoma azul presenta expresión membranosa de ese marcador. La OMS considera que su riesgo de transformación a melanoma es bajo/intermedio.
Aunque la OMS aconseja una escisión completa con márgenes amplios (5mm) para aquellos tumores melanocíticos de alto riesgo de progresión a melanoma, no todos los autores están de acuerdo en que el riesgo de progresión de los distintos melanocitomas sea un hecho probado51.
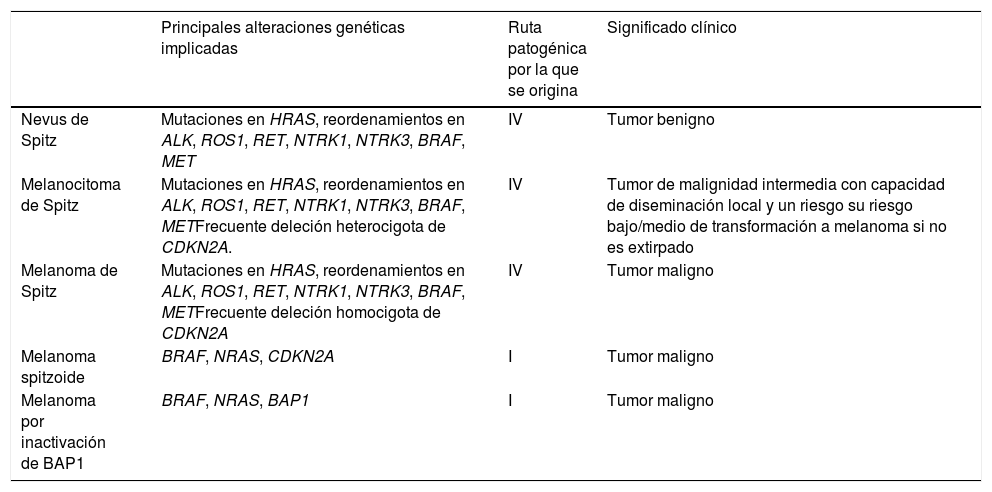
Un melanoma spitzoide no es lo mismo que un melanoma de Spitz, ni un tumor de Spitz lo mismo que un tumor spitzoide (tabla 2)El melanoma de Spitz es el punto final de la ruta patogénica IV que se inicia en los nevus de Spitz y pasa por el melanocitoma de Spitz.
Diferencias entre los principales tumores melanocíticos con fenotipo «spitzoide»
| Principales alteraciones genéticas implicadas | Ruta patogénica por la que se origina | Significado clínico | |
|---|---|---|---|
| Nevus de Spitz | Mutaciones en HRAS, reordenamientos en ALK, ROS1, RET, NTRK1, NTRK3, BRAF, MET | IV | Tumor benigno |
| Melanocitoma de Spitz | Mutaciones en HRAS, reordenamientos en ALK, ROS1, RET, NTRK1, NTRK3, BRAF, METFrecuente deleción heterocigota de CDKN2A. | IV | Tumor de malignidad intermedia con capacidad de diseminación local y un riesgo su riesgo bajo/medio de transformación a melanoma si no es extirpado |
| Melanoma de Spitz | Mutaciones en HRAS, reordenamientos en ALK, ROS1, RET, NTRK1, NTRK3, BRAF, METFrecuente deleción homocigota de CDKN2A | IV | Tumor maligno |
| Melanoma spitzoide | BRAF, NRAS, CDKN2A | I | Tumor maligno |
| Melanoma por inactivación de BAP1 | BRAF, NRAS, BAP1 | I | Tumor maligno |
Esta ruta conlleva alteraciones genéticas típicas de la «familia Spitz», como la ausencia de mutaciones en BRAF, en NRAS o en NF1, y frecuente presencia de reordenamientos en los genes de las cinasas o mutaciones HRAS. Como las cinasas no suelen expresarse en melanocitos maduros normales y como la fusión de cinasas suele conllevar su expresión, estas fusiones pueden detectarse por inmunohistoquímica, mediante el uso de anticuerpos contra ellas. Sin embargo, se debe tener en cuenta que no todas las fusiones son igual de efectivas en propiciar la expresión de la cinasa correspondiente. Sobre todo, por lo que respecta a ALK y a ROS1, la positividad en inmunohistoquímica puede ser variable, yendo desde citoplásmica difusa a moteada (dot-like). De hecho, la tinción con ROS1 es frecuentemente débil. En el caso de NTRK (1 y 3), la expresión suele ser tan intensa, que expresiones moderadas, débiles o parcheadas, deberían ser consideradas dudosas por varios motivos: 1) porque hay isoformas de NTRK que pueden expresarse en melanocitos normales, y 2) porque NTRK puede expresarse en tumores melanocíticos no Spitz (como nevus azules o nevus penetrantes profundos), debido a sobreexpresión de ALK mediante otros mecanismos moleculares. También se debe tener en cuenta la expresión nuclear de NTRK, que suele verse más en Spitz con fusiones de NTRK3 que en los de NTRK1. En la evaluación inmunohistoquímica de la expresión de las cinasas no se debe malinterpretar como positiva la tinción de los macrófagos. Para otras cinasas (BRAF, RAF1 o MAP3K8), la inmunohistoquímica no parece servir para predecir la existencia de fusiones36.
Existen tumores melanocíticos (tanto benignos como malignos) con rasgos spitzoides en el estudio con hematoxilina eosina que no son, sin embargo, tumores de Spitz, porque no siguen la ruta patogénica de los Spitz y, por lo tanto, no muestran las alteraciones genéticas características de estos. Un ejemplo serían los tumores spitzoides en adultos que siguen la ruta patogénica I y que frecuentemente muestran mutaciones en BRAF, NF1 o NRAS. De hecho, y puesto que la transformación maligna de un tumor de Spitz benigno en un melanoma es un fenómeno raro, los melanomas de Spitz no son frecuentes. Son mucho más habituales los melanomas con aspecto spitzoide.
De entre los melanomas spitzoides, hay un grupo que requiere mención especial: se trata de los que presentan inactivación de BAP1. No hace mucho, estas lesiones melanocíticas se consideraban erróneamente en el espectro de los tumores de Spitz. No dejaba por ello de sorprender, que en su mayoría mostrasen mutaciones de BRAF o de RAF167, algo perfectamente entendible en el presente, ya que sabemos que estos melanomas pertenecen a la ruta patogénica I, de baja exposición solar, y las mutaciones en BAP1 son un evento más en la transformación a melanoma. Por eso en el examen con hematoxilina eosina, la mayoría de los BAPomas corresponden a nevus combinados, con un nódulo focal del melanocitoma con mutación en BAP1, en el seno de un nevus común con mutaciones en BRAF. El nódulo del melanocitoma comparte la misma mutación en BRAF que el resto de los nevus debido a que la mutación en BAP1ha sido posterior a la de BRAF. A veces estos tumores muestran aspecto de tumor spitzoide, pero con infiltrado inflamatorio prominente, por lo que en el pasado, muchos fueron clasificados de nevus de Spitz en halo. Sin embargo, no todos los tumores melanocíticos spitzoides con prominente infiltrado son BAPomas. Así, verdaderos tumores de Spitz con fusiones en NTRK1 pueden tener gran cantidad de linfocitos acompañantes.
Un tumor de Spitz atípico no es un melanocitoma de Spitz y, por lo tanto, no deberían ser términos intercambiablesPor el mismo motivo, el melanocitoma de Spitz pertenece a la ruta patogénica de la familia Spitz. Por consenso, un melanocitoma de Spitz debe mostrar deleción del CDKN2A con negatividad para p16 pero en ausencia de aberraciones cromosómicas numéricas adicionales y sin otras mutaciones adicionales, especialmente en el promotor del TERT. Aunque los términos melanocitoma de Spitz y tumor de Spitz atípico se han intercambiado en muchas ocasiones en la literatura, el término melanocitoma de Spitz debería ser reservado para aquellas lesiones con las alteraciones genéticas comentadas.
Por otro lado, muchos de los nevus que en el pasado fueron clasificado como tumores de Spitz atípicos son hoy reconocidos como otros tipos de tumores melanocíticos incluidos en otras rutas patogénicas, como la I (de baja exposición solar), dadas sus alteraciones en BRAF, NRAS o BAP1.
El concepto de nevus de sitio especialAlgunos nevus melanocíticos muestran características atípicas o preocupantes en el estudio con hematoxilina eosina. Sin embargo, los estudios genético y molecular de estos nevus no han revelado resultados de riesgo en términos pronósticos o de progresión a malignidad. Esto hace pensar que las alteraciones histológicas son resultado de la localización de dichos nevus en zonas topográficas especiales (zonas de roce o exposición a otros agentes, por ejemplo)68.
Los principales «sitios especiales» son la mama y los pliegues corporales, el cuero cabelludo, el área genital y las zonas acrales68.
Las alteraciones histológicas evidenciadas en estos nevus son variadas pero muchas veces preocupantes e incluyen confluencia de tecas, cierto grado de extensión pagetoide, áreas de fibrosis dérmica, infiltrados inflamatorios, pérdida de cohesión melanocítica en las tecas, patrón irregular de distribución de las tecas o hipercromasia nuclear melanocítica. El conocimiento de las posibles alteraciones asociadas a cada localización anatómica es mandatorio para no realizar un sobrediagnóstico de melanoma en estos casos.
Otra de las importantes consideraciones sobre estos nevus de sitio especial es que, a pesar de mostrar atipia no son «nevus displásicos», entendiendo por tal el concepto de nevus marcador del riesgo de melanoma en algunas familias69 y, por lo tanto, no deberían ser diagnosticados como tales.
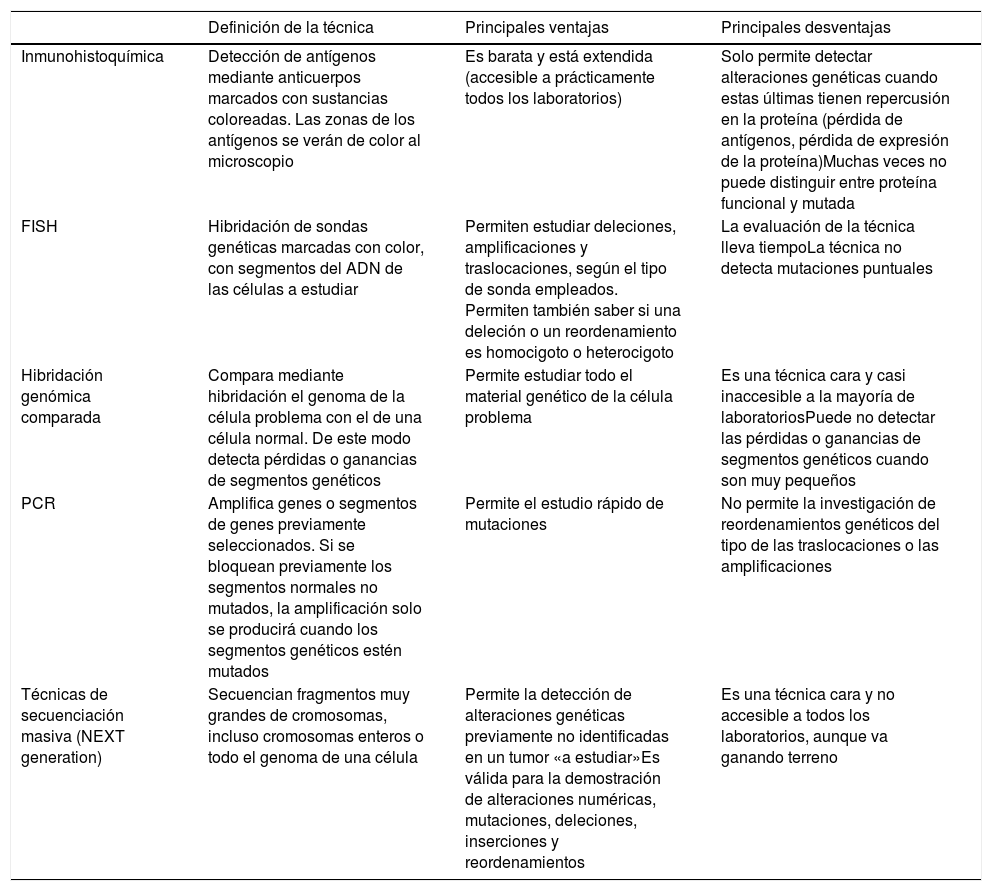
Principales técnicas de detección de alteraciones genéticas usadas en la práctica diaria (tabla 3)Los tumores melanocíticos muestran alteraciones genéticas desde su inicio, es decir, incluso cuando son benignos. También pueden existir alteraciones epigenéticas que inactiven al gen, sin necesidad de que haya alteraciones estructurales. Estas últimas no podrían ser, por lo tanto, detectadas por las técnicas genómicas habitualmente empleadas. Las alteraciones genéticas son variadas, y van, por ejemplo, desde mutaciones puntuales hasta pérdidas de segmentos cromosómicos más o menos grandes, brazos completos, cromosomas enteros o duplicaciones de material genético (partes de cromosomas o cromosomas enteros).
Principales técnicas complementarias empleadas en el estudio de los tumores melanocíticos
| Definición de la técnica | Principales ventajas | Principales desventajas | |
|---|---|---|---|
| Inmunohistoquímica | Detección de antígenos mediante anticuerpos marcados con sustancias coloreadas. Las zonas de los antígenos se verán de color al microscopio | Es barata y está extendida (accesible a prácticamente todos los laboratorios) | Solo permite detectar alteraciones genéticas cuando estas últimas tienen repercusión en la proteína (pérdida de antígenos, pérdida de expresión de la proteína)Muchas veces no puede distinguir entre proteína funcional y mutada |
| FISH | Hibridación de sondas genéticas marcadas con color, con segmentos del ADN de las células a estudiar | Permiten estudiar deleciones, amplificaciones y traslocaciones, según el tipo de sonda empleados. Permiten también saber si una deleción o un reordenamiento es homocigoto o heterocigoto | La evaluación de la técnica lleva tiempoLa técnica no detecta mutaciones puntuales |
| Hibridación genómica comparada | Compara mediante hibridación el genoma de la célula problema con el de una célula normal. De este modo detecta pérdidas o ganancias de segmentos genéticos | Permite estudiar todo el material genético de la célula problema | Es una técnica cara y casi inaccesible a la mayoría de laboratoriosPuede no detectar las pérdidas o ganancias de segmentos genéticos cuando son muy pequeños |
| PCR | Amplifica genes o segmentos de genes previamente seleccionados. Si se bloquean previamente los segmentos normales no mutados, la amplificación solo se producirá cuando los segmentos genéticos estén mutados | Permite el estudio rápido de mutaciones | No permite la investigación de reordenamientos genéticos del tipo de las traslocaciones o las amplificaciones |
| Técnicas de secuenciación masiva (NEXT generation) | Secuencian fragmentos muy grandes de cromosomas, incluso cromosomas enteros o todo el genoma de una célula | Permite la detección de alteraciones genéticas previamente no identificadas en un tumor «a estudiar»Es válida para la demostración de alteraciones numéricas, mutaciones, deleciones, inserciones y reordenamientos | Es una técnica cara y no accesible a todos los laboratorios, aunque va ganando terreno |
Con la técnica de FISH logramos hibridaciones de sondas coloreadas y segmentos cromosómicos del tumor melanocítico. De ese modo, podemos detectar ausencia, cambio de posición (traslocaciones, fusiones, etc.) o amplificaciones de tales segmentos. Hay 3sondas especiales que merecen mención en el diagnóstico de tumores melanocíticos de difícil filiación70,71: sondas para las regiones CCND1 (11q13), RREB1 (6p25) y MYB (6q23). La combinación de las 3ha dado buenos resultados en la distinción entre melanomas y nevus. Los resultados deben siempre ser contextualizados con los hallazgos histopatológicos. Así, por ejemplo, una sola traslocación de las mencionadas no es sinónimo de melanoma. De igual manera, un test negativo para varias sondas tampoco es garante absoluto de benignidad.
Con la hibridación genómica comparada (HGC), el genoma del tumor melanocítico es comparado con el de una célula normal adyacente a la lesión72. De ese modo se detectan pérdidas o ganancias de segmentos cromosómicos más o menos grandes, e incluso de cromosomas enteros. Solo cuando los segmentos son demasiado pequeños pueden quedar «invisibles» a la HGC. Mientras que los nevus no suelen tener alteraciones en las copias de segmentos cromosómicos (con algunas excepciones, como las amplificaciones de 11p vistas en algunos nevus de Spitz), los melanomas suelen presentar muchas alteraciones numéricas, que incluyen segmentos de cromosomas o cromosomas completos.
La PCR es una técnica de amplificación de segmentos cortos genómicos que permite detectar mutaciones en un determinado gen. Se basa en el principio siguiente: mediante un polímero sintético similar al ADN, se bloquean los segmentos normales del gen a estudiar, de tal manera que solo se amplificaran en los ciclos de PCR aquellos segmentos mutados. Esta técnica da muy buenos resultados para la detección de mutaciones en BRAF y NRAS73.
El resultado óptimo final de la transcripción y la traducción de un gen es la producción de proteína. Las proteínas pueden detectarse mediante inmunohistoquímica, que usa anticuerpos contra antígenos proteicos. De esta manera, por ejemplo, detectamos las cinasas citoplásmicas de muchos tumores de Spitz36. También tiene valor la pérdida de expresión de proteína, que significa negatividad en inmunohistoquímica. Las limitaciones de esta técnica son aquellos casos de producción de proteínas anómalas no funcionales, en los que la inmunohistoquímica seguiría siendo positiva. Dentro de la inmunohistoquímica, merece mención especial el anticuerpo PRAME (del inglés PReferentially expressed Antigen in MElanoma), cuya expresión nuclear debe verse con preocupación ante una lesión melanocítica histológicamente atípica74,75. La tinción con PRAME no debe evaluarse en términos absolutos (positividad/negatividad), sino relativos (intensidad y porcentaje de tinción), y debe siempre ser contextualizada con los hallazgos histológicos. Usada de esa manera ha contribuido mucho a la orientación de muchas lesiones melanocíticas en la práctica diaria. Uno de sus mayores inconvenientes son, sin embargo, los tumores melanocíticos de morfología spitzoide, donde los resultados con PRAME no son tan buenos como en otros tipos de tumores melanocíticos. En este sentido, recientemente, sin embargo, hay un artículo reciente que ha presentado la combinación de la inmunohistoquímica para p16 y BRAF V600, como superior a PRAME en tumores de fenotipo spitzoide76.
Complementariamente a las técnicas de secuenciación de segmentos genéticos cortos, se han desarrollado técnicas de secuenciación masiva, que permiten la secuenciación de fragmentos muy grandes de ADN. Además de poder estudiar el genoma entero, permiten el estudio selectivo del exoma, o de unos cuantos genes seleccionados. Esto permite detectar alteraciones genéticas incluso no descritas en un tumor concreto. Las secuenciaciones de nueva generación (next generation sequencing [NGS]) combinan secuenciaciones de material ingente de ADN con unos costes asequibles para el uso diagnóstico diario. La NGS es capaz de demostrar alteraciones numéricas, mutaciones, deleciones, inserciones y reordenamientos. Esta técnica ha sido usada con éxito en la categorización y diagnóstico del melanoma77.
ConclusionesEn los últimos años se ha impuesto la evidencia de que los tumores melanocíticos malignos se desarrollan en rutas que implican alteraciones genéticas y epigenéticas acumulativas y que van desde precursores benignos, de malignidad intermedia, o formas malignas in situ, hasta el polo final maligno del espectro. Hasta la fecha, han sido identificadas 9rutas patogénicas que acaban en algún tipo de melanoma. Algunas de estas rutas muestran una gran relación con exposición solar mientras que otras no están relacionadas con el sol. Cada ruta muestra unas alteraciones genéticas características identificables que nos han resultado cruciales para el diagnóstico correcto cada tumor melanocítico. La histopatología con hematoxilina-eosina sigue, no obstante, representando un papel diagnóstico fundamental y es el escenario en el que todas las demás técnicas auxiliares usadas, se deben contextualizar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



