




Actualmente existe un incremento del número de casos de patología oral en los servicios de Dermatología. Dentro de las diversas lesiones que forman parte de esta amplia área de conocimiento destacamos la leucoplasia oral, por ser la lesión mucosa precancerosa más frecuente y la que mayores problemas origina tanto en su diagnóstico como en su manejo terapéutico.
En esta revisión, además de definir la leucoplasia, establecemos un diagnóstico diferencial con el resto de las patologías orales más importantes y analizamos los diversos parámetros clínicos, histológicos y moleculares que definen el riesgo de transformación maligna de esta patología. Finalmente se establece un algoritmo terapéutico que nos ayuda a optimizar su manejo clínico.
Dermatology departments are currently seeing an increase in the number of cases of oral diseases. Of note among the range of lesions covered by this broad area of knowledge is oral leukoplakia—the most common precancerous lesion and the most problematic in terms of both diagnosis and therapeutic management.
In addition to defining leukoplakia, this review also establishes a differential diagnosis with the other most important oral diseases and analyzes the different clinical, histologic, and molecular features that can provide an indication of the risk of malignant transformation. Finally, a therapeutic algorithm is presented to help us optimize clinical management of the disease.
El cáncer oral es la neoplasia más frecuente de cabeza y cuello, con una incidencia mundial que excede los 300.000 casos anuales. Se trata de una neoplasia que causa una importante morbimortalidad, con una supervivencia a los 5 años menor del 50 %1,2. Entre las nuevas perspectivas para el control de este cáncer se incluye la detección precoz de la leucoplasia, considerada como la lesión premaligna oral más común de la cavidad oral, que aparece hasta en el 60 % de los pacientes diagnosticados de carcinoma oral de células escamosas (CE)3, y cuya presencia supone un marcador de aumento del riesgo de cáncer orofaríngeo 4.
Definición de leucoplasiaEl término leucoplasia oral hace referencia a una entidad clínica definida por la Organización Mundial de la Salud (OMS) como«lesión predominantemente blanquecina localizada en la mucosa oral que no puede ser caracterizada como ninguna otra lesión definida ni desde el punto de vista clínico ni histológico»5. En la práctica diaria la definición se completa incluyendo el dato de la imposibilidad de la eliminación de la lesión mediante el simple rascado, lo cual facilita su distinción frente a la candidiasis pseudomembranosa 6. Tiene una prevalencia que oscila entre el 0,4 y el 0,7 % de la población. La distribución por sexos es variable y tiene mayor incidencia en pacientes que presentan hábitos tóxicos tales como el tabaco y/o el alcohol 7.
La importancia de estas lesiones radica en su capacidad de transformación en un CE, porcentaje que oscila entre un 3 y un 17,5 %. Entre un 16 y un 62 % de los CE se han asociado a la presencia de leucoplasia oral en el momento del diagnóstico, por lo que se trata de una entidad estrechamente asociada al cáncer de cabeza y cuello 8-10. Estas diferencias en los porcentajes de malignización se justifican por los diferentes criterios utilizados para definir la leucoplasia, y por la diversidad geográfica, asociada a diferentes hábitos sociales y variaciones genéticas.
La leucoplasia, junto con la eritroplasia, el liquen plano y la fibrosis submucosa, forma parte del conjunto de lesiones orales precancerosas. La prevención de la transformación maligna es crítica, ya que la tasa de supervivencia a los 5 años tras el diagnóstico de CE es de un 50 %1,2.
El estudio comparativo entre aquellas leucoplasias que no malignizan a largo plazo frente a aquellas que sí lo hacen ha permitido definir una serie de características clínicas, histológicas y moleculares que definen un grupo de leucoplasias conocido como«de alto riesgo de malignización». La posibilidad de detectar esta variante permitiría establecer unos controles evolutivos más estrechos y una terapéutica más agresiva.
Etiología de la leucoplasiaEl establecimiento de un factor etiológico para una lesión blanquecina excluye el diagnóstico de leucoplasia, excepto en el caso del tabaco. Las infecciones por Candida, papilomavirus (VPH) y recientemente por virus de Epstein Barr (VEB) son cofactores que pueden modificar el pronóstico evolutivo de la leucoplasia ya establecida.
Leucoplasia y tabacoEl tabaco es un potente carcinógeno reconocido como el factor de riesgo más importante para el desarrollo de carcinomas de cabeza y cuello, junto al etilismo crónico 1,2. Por ello, ante un paciente con leucoplasia oral y hábito tabáquico hay que diferenciar entre la denominada estomatitis nicotínica o«paladar del fumador», y la verdadera leucoplasia.
La estomatitis nicotínica o«el padadar del fumador»se corresponde al cuadro clínico caracterizado por lesiones blanquecinas localizadas en la superficie del paladar blando o en el suelo de la boca, en la que el tabaco es el agente causal directo 11 (tabla 1). Dicha relación causal se puede objetivar por su resolución completa tras la suspensión del hábito tabáquico durante un intervalo de 4 a 6 meses. Una característica clínica típica de este cuadro es la existencia de un«patrón en huella dactilar»o«en piedra pómez», que se caracteriza por la presencia de una estriación fina blanquecina en la superficie, que imita a una huella dactilar 12. Típicamente estas lesiones presentan un patrón histológico de queratinización«en árbol de navidad», «en campanario de iglesia»o«en v». Son lesiones sin potencial maligno, que se producen por la actividad proliferativa provocada por el tabaco a través de la inducción del receptor del factor de crecimiento epidérmico (EGFR), que activa de forma secundaria a la ciclina D1 12.
Datos anatomopatológicos que definen un epitelio oral displásico
| Aumento del volumen celular y nuclear y pleomorfismo |
| Hiperplasia de las células basales |
| Presencia de núcleos hipercromáticos |
| Nucleolos agrandados prominentes |
| Aumento de la proporción núcleo: citoplasma |
| Queratinización prematura de células independientes |
| Aumento del índice mitótico |
| Mitosis por encima de la capa basal |
| Pérdida de polaridad celular |
| Disminución de la adhesión celular |
| Crestas epidérmicas bulbosas/fusionadas |
Sin embargo, el mantenimiento de este carcinógeno a largo plazo puede provocar un aumento de la frecuencia de aparición de mutaciones en la mucosa oral y, de manera indirecta, la inestabilidad genómica 6, que clínicamente se manifestaría como la pérdida del patrón en huella dactilar y la no resolución de la lesión tras la retirada del tabaco. En este caso la lesión cumpliría los requisitos de la definición de la leucoplasia.
Leucoplasia y CandidaLa implicación de especies de Candida en la etiología o en la progresión de las leucoplasias es controvertida. Diversos tipos de Candida productores de nitrosaminas (diferentes a la variante albicans común) se han aislado de leucoplasias clínicamente heterogéneas con displasia histológica 13,14. La erradicación de esta micosis superficial no conlleva la resolución de la lesión. Sin embargo, la eliminación del hongo sí provoca la transformación de la variante heterogénea de alto riesgo de la leucoplasia en la forma homogénea de bajo riesgo. Por ello se considera a esta sobreinfección fúngica como un factor prooncogénico relevante 15.
Leucoplasia y papilomavirusLa posible implicación del VPH en la etiología y en el potencial de transformación maligna de las lesiones orales premalignas ha sido estudiada extensamente, dada la gran importancia de este agente en el cáncer ginecológico del cérvix uterino 16-18. Sin embargo, la implicación como agente etiológico en el desarrollo de la leucoplasia o como una simple sobreinfección es debatida. Los VPH más estudiados mediante técnicas de hibridación in situ incluyen los tipos 6, 11, 16, 18, 31, 33 y 35. Existe una asociación bien establecida entre la infección por VPH16 y 18 y el desarrollo de CE de cérvix 19. Sin embargo, existe controversia en la existencia de asociación en la región oral. En un reciente estudio se detecta una incidencia de VPH de 2 a 3 veces superior en lesiones precancerosas orales y de 4–5 veces en CE que en epitelio normal 19. Los VPH de alto riesgo están más frecuentemente asociados con el CE que los de bajo riesgo, por ello se concluye que la infección por VPH16 supone un factor de riesgo para malignización de la leucoplasia independiente de los efectos nocivos del tabaco y el alcohol sobre la mucosa oral.
La variante verrugosa proliferativa ha sido asociada en múltiples ocasiones a una coinfección por VPH, en especial con los serotipos 16 y 18. Ello se basa en el potencial oncogénico de estos VPH en otras mucosas y en la evolución rápida de esta variante clínica. Sin embargo, la ausencia de detección mediante reacción en cadena de la polimerasa (PCR) del VPH en un estudio prospectivo realizado por Bagán et al 20 sobre un total de 13 leucoplasias verrugosas proliferativas pone en seria duda su relación patogénica.
Leucoplasia y virus de Epstein-BarrEl VEB ha sido implicado en un gran número de neoplasias malignas, incluyéndose en algunos estudios como factor etiológico del CE oral.
En un reciente estudio preliminar no se ha encontrado evidencia de asociación causal entre la leucoplasia verrugosa proliferativa y el VEB; sin embargo, dicho estudio fue realizado sobre una pequeña muestra de pacientes, por lo que no se puede descartar su asociación etiopatogénica 21.
Patogenia de la leucoplasiaUn mejor conocimiento de la biología molecular y del proceso de desarrollo del cáncer es la única vía para optimizar nuestras posibilidades de predecir el potencial oncogénico de la leucoplasia. Recientemente, mediante estudios de biología molecular, se ha descrito que un porcentaje variable de leucoplasias orales presenta alteraciones moleculares en común con el CE oral, con potencial oncogénico independiente de la atipia histológica. De hecho, la aparición de estas alteraciones citogenéticas se ha descrito en leucoplasias sin atipia celular.
Pérdida de heterocigosidad o inestabilidad de microsatélitesLa pérdida de heterocigosidad (PDH) en una célula representa la pérdida de la función normal de un alelo de un gen cuyo alelo homólogo estaba previamente inactivado. Esta inactivación previa ocurre en las células germinales parentales, y es transmitida a sus descendientes generándose células heterocigotas para dicho gen.
El desarrollo de este fenómeno en regiones cromosómicas que contienen genes supresores tumorales podría estar relacionado con el proceso de malignización 22. La presencia de PDH en la leucoplasia oral y su posible valor predictivo han sido recientemente revisados por Zhang y Rosin 23, que establecen que las lesiones con PDH limitada a los cromosomas 3p y/o 9p formarían parte del grupo de leucoplasias de riesgo intermedio, con un incremento de riesgo de malignización de 3,8 veces, mientras que aquellas lesiones que asocian la PDH de los cromosomas 3p y/o 9p con la pérdida de uno o más de los siguientes cromosomas: 4q, 8p, 11q, 13q y 17p, serían consideradas como leucoplasias de alto riesgo, con un riesgo hasta 33 veces superior de progresión a cáncer. Por último, se considerarían leucoplasias de bajo riesgo aquellas que no presentan ninguna PDH de las mencionadas.
El estudio de estas alteraciones moleculares en los márgenes de resección complementa la información histológica, y puede demostrar la existencia de alteraciones moleculares en bordes histológicamente libres de enfermedad, lo que nos justificaría las recurrencias y el desarrollo de CE oral 24. De hecho, la confirmación molecular de la resección completa de la lesión precancerosa se mostró altamente correlacionada con la reducción de riesgo de carcinoma oral en aquellos pacientes con leucoplasias de riesgo alto e intermedio, hecho que no ocurrió en aquellas formas de bajo riesgo 25.
AneuploidiaEl contenido de ADN, o ploidia de ADN, informa del grado de inestabilidad genética y de aberraciones de la secuencia genómica. En el caso de las neoplasias, las células diploides genéticamente estables son sustituidas por células aneuploides inestables. Esta alteración ha sido estudiada mediante técnicas de citometría de flujo en los CE de cavidad oral. Los estudios realizados por Sudbø et al 24 se focalizaron en la medida del estado de ploidia en las leucoplasias orales con displasia histológica. En este trabajo se comprobó que la aneuploidia en la leucoplasia displásica era un marcador pronóstico de progresión a carcinoma, con independencia de la resección con bordes histológicamente libres de enfermedad. Así, de las lesiones displásicas analizadas, un 70 % fueron lesiones diploides que se consideraron de bajo riesgo (el 3 % evolucionaron a CE), un 13 % fueron lesiones tetraploides de riesgo intermedio (el 60 % progresaron a CE), mientras que las lesiones aneuploides fueron consideradas la variante de alto riesgo. Dicho grupo supuso un 17 % del total de las lesiones displásicas, con una tasa de malignización del 84 %. A pesar de que los resultados obtenidos por Sudbø han sido puestos en duda 26, son necesarios nuevos estudios para determinar el valor pronóstico de este prometedor marcador.
p53La mutación de p53, gen supresor tumoral, representa la anomalía genética más común en el cáncer del ser humano 27. Su función fisiológica es la de prevenir la acumulación de daño genético celular, bien reparando el daño previamente a la división celular, bien causando la muerte celular por apoptosis. La p53 desempeña un papel fundamental en la reparación del ADN y en la regulación del ciclo celular después del daño inducido por carcinógenos en el ADN del epitelio oral. La p53 se activa mediante fosforilación de sus residuos serina como respuesta al estrés celular inducido por carcinógenos, de forma que el contenido celular de proteína p53 activa determina la respuesta que sigue al daño inducido por carcinógenos en el ADN. Cuando el contenido celular de p53 es bajo o intermedio, las células evolucionan a la detención del ciclo celular para permitir la reparación del ADN. Sin embargo, si el contenido celular de p53 es alto, las células entran en apoptosis. La existencia de una mutación en el gen p53 conlleva un fracaso en la vía protectora de la célula, pudiéndose producir la transformación neoplásica en el epitelio. De hecho, se ha demostrado que la presencia de una mutación en la p53 hace que después de la irradiación con UVB de los queratinocitos, éstos entren en apoptosis. Sin embargo, cuando la p53 acumula dos o más mutaciones se produce la transformación neoplásica.
La p53 natural tiene una vida media corta y suele presentarse en el interior de los queratinocitos epidérmicos en tan escasa cantidad que no son detectables en la inmunohistoquímica. Por el contrario, la forma mutante de p53 presenta una vida media más larga, lo que provoca su acumulación en el interior de las células epidérmicas, permitiendo su visualización mediante tinciones inmunohistoquímicas.
La marcada inespecificidad mostrada por la mutación de la p53 como factor pronóstico de malignidad está probablemente relacionada con la imposibilidad de detectar la variante mutante de p53 de manera selectiva mediante la prueba inmunohistoquímica 28,29. Así, ese incremento de la positividad en la tinción inmunohistoquímica para p53 puede representar un aumento de p53 no mutante como respuesta a agresiones externas. Por ello, sería recomendable la realización combinada de estudios inmunohistoquímicos y moleculares cuando se pretenda establecer el valor pronóstico de esta mutación.
Poeta et al 30 realizaron un análisis de la presencia de mutaciones de p53 en el CE de cabeza y cuello mostrando una prevalencia en un 53 % de los pacientes. En este estudio se apreció una asociación estadísticamente significativa entre mutaciones disruptivas del p53 y un descenso de la supervivencia después del tratamiento quirúrgico del CE de cabeza y cuello, estableciéndose dicha mutación como un factor de riesgo independiente de mal pronóstico.
Actividad telomerasa en la leucoplasiaLa telomerasa es una enzima formada por un complejo proteína-ácido ribonucleico con actividad polimerasa, que es producida en las células germinales embrionarias y cuya función es el alargamiento de los telómeros mediante la realización de copias de la secuencia TTAGGG. Esta proteína desempeña un papel importante en la formación, mantenimiento y renovación de los telómeros, impidiendo la apoptosis celular. Es reprimida por las células somáticas maduras tras el nacimiento, permitiendo el acortamiento del telómero después de cada división celular. La sobreexpresión de telomerasa se ha descrito asociada a diversos procesos neoplásicos31.
El gen hTERT codifica la subunidad catalítica de la telomerasa y muestra una correlación positiva con la actividad de la telomerasa en diversos estudios moleculares. Su sobreexpresión en la leucoplasia, que traduce un aumento de la actividad telomerasa, representa un fenómeno precoz en el proceso de la carcinogénesis oral, que puede ser detectado en etapas precancerosas. Este fenómeno muestra una marcada correlación positiva con el grado de atipia, mostrando cambios displásicos graves 32.
Análisis mediante micromatrizEl desarrollo de esta técnica permite realizar un cribado de todo el genoma y se utiliza para comparar lesiones displásicas y tejido sano. Así, los genes sobrerregulados en las lesiones displásicas incluyen los genes relacionados con la inflamación (ciclooxigenasa 2 [COX-2], variante decorin A2, ALOX5, ALOX12, PTGES), ciertos genes de receptores (PTGER, 3b1, a2)33 y ciertos marcadores genéticos asociados a malignidad (psorasin [PSOR1] y versican [CSPG2]) 34.
Marcadores tisularesCarbohidratos de la superficie celularLos carbohidratos de superficie con actividad antigénica de grupo sanguíneo están ampliamente distribuidos en los diferentes tejidos humanos. Estos marcadores, que incluyen los antígenos del sistema ABO, Lewis y T/Tn, se presentan en la superficie de las células epiteliales del epitelio escamoso oral 32.
Durante el desarrollo tumoral maligno, la síntesis de estos carbohidratos se altera 33 por la expresión aberrante de las glucosiltransferasas. Así, en las displasias epiteliales se detecta una pérdida de los antígenos de histocompatibilidad sanguíneos (A o B) en el estrato espinoso, así como un aumento en el número de estratos celulares teñidos para su precursor molecular (H-antígeno), que normalmente se expresa únicamente en las células parabasales. Algunos patrones de expresión aberrante aparecen en lesiones premalignas sin displasia epitelial, lo que sugiere que los cambios de antígeno de histocompatibilidad sanguíneo aparecen en etapas precoces del desarrollo tumoral.
Estos cambios de expresión de los antígenos de superficie celular aparecen en lesiones premalignas y malignas. Sin embargo, también son detectados en lesiones benignas, como en el proceso de reparación de heridas 35, lo cual puede dificultar la interpretación de los resultados.
En determinados tumores, como en el cáncer de cérvix, el cáncer de cabeza y cuello y los carcinomas de la cavidad oral, la expresión de estos grupos sanguíneos de histocompatibilidad en las células tumorales ha mostrado una implicación pronóstica. De esta forma, la pérdida de la expresión de los antígenos A o B aumenta la motilidad de las células tumorales, su invasión en matrigel y su potencial tumoral en animales singénicos 36,37.
QueratinasSon proteínas que constituyen el citoesqueleto de las células epiteliales. Actualmente se conocen 20 queratinas, que se numeran de 1 a 20. En el epitelio escamoso oral se detecta un grupo de queratinas que se presentan en circunstancias normales. Sin embargo, durante el proceso de malignización se producen cambios en el tipo y en la distribución de estas proteínas, por lo que pueden representar otro marcador potencial de progresión displásica a CE.
En todo epitelio oral normal la pareja de queratinas K5/K14 se encuentra en la membrana basal, mientras que K4/K13 y K1/K10 están presentes en el estrato espinoso del epitelio no cornificado y cornificado, respectivamente 36. En el epitelio displásico se producen una serie de cambios: la K5/K14 se expresa en el estrato parabasal y en el estrato espinoso, así como en su localización habitual, probablemente reflejando una hiperplasia de las células basales. Por otro lado, la expresión de K4/K13 y K1/K10 se encuentra reducida e incluso ausente. Estos cambios muestran una marcada correlación con el grado de displasia histológica 37,38.
En el epitelio oral normal K8/K18 se expresan en el estrato basal y espinoso suprabasal del epitelio simple y generalmente no son detectadas mediante inmunohistoquímica. Sin embargo, en la displasia epitelial oral estas queratinas son detectadas mediante tinción inmunohistoquímica en más de la mitad de los casos.
La K19 está presente de manera natural en el estrato basal de epitelio oral no cornificado, pero no en aquellos epitelios cornificados. En la displasia grave, sin embargo, existe una fuerte expresión de K19 tanto en estratos basales como suprabasales de epitelios cornificados o no cornificados. Esta última queratina también puede aparecer sobreexpresada en procesos inflamatorios gingivales, suponiendo una limitación en el estudio de su posible valor pronóstico en los epitelios displásicos 39,40.
Esta pérdida de la diferenciación basada en la expresión de queratinas en lesiones displásicas puede correlacionarse con un mayor riesgo de transformación a CE, pero actualmente no existen estudios relacionados.
IntegrinasSon receptores de superficie celular que median los mecanismos de coordinación célula-célula y célula-matriz extracelular. Estos receptores están compuestos de 22 subunidades α y β, cada una con una especificidad de unión a ligando. El epitelio basal normal expresa integrinas α2p1 y α3β1. La expresión de αpβ se encontró en el 90 % de los CE y en el 27 % de las leucoplasias displásicas estudiadas, pero no en piel normal. Esta integrina estaba asociada a una transformación de las lesiones epiteliales displásicas. Sin embargo, su expresión no es suficiente de manera aislada para la progresión de displasia a CE 38.
La familia de glucoproteínas CD44 está también relacionada con las interacciones célula-célula y célula-matriz. Su isoforma CD44v7-8 presenta una expresión disminuida en el CE respecto a leucoplasias displásicas. A su vez, los tumores con expresión de esta isoforma presentan una supervivencia a los 5 años mayor que aquéllos con la isoforma negativa, sugiriendo que su pérdida se asocia con peor pronóstico 39.
Receptor del factor estimulante de colonias granulocíticasSu expresión está aumentada en la displasia y en el CE frente a epitelio normal e hiperplásico. Probablemente esto sea la expresión de un intento de bloqueo del defecto de maduración, bien en la fase inicial precancerosa, bien en el cáncer establecido40.
Receptores de factor de crecimientoLa presencia de una sobreexpresión de factor de crecimiento transformante (TGF)-α, tanto en términos de área como de intensidad de la tinción, se presenta de forma paralela a la gravedad de la displasia oral. De la misma manera, el EGFR mostró un incremento en la intensidad de la tinción de forma paralela al aumento de la displasia41.
Reguladores del ciclo celularLa pérdida de la expresión de p16 expresado por el gen 9p21, considerado el gen que más precozmente se inactiva en el CE de cabeza y cuello, precede a los cambios histológicos en la mucosa oral. A su vez, se advirtió un incremento secuencial en la expresión de la ciclina D1, observada desde tejido normal a hiperplásico, displásico y finalmente CE42. Junto a ello se pudo constatar que el gen de la ciclina D1 estaba amplificado en un 70 % de leucoplasias que progresaban a CE43.
Metaloproteinasas de la matrizComprenden un total de 20 enzimas proteolíticas que desempeñan un papel esencial en el remodelado tisular. Se ha detectado un aumento en los niveles de metaloproteinasas 1 y 9 en aquellas displasias que progresan a CE frente a aquellas que no lo hacen44.
EMMPRIN (CD147 y M6)Se trata de un inductor de actividad de las metaloproteinasas cuya expresión parece estar correlacionada positivamente con el grado de displasia. Sin embargo, dicha expresión disminuye en las células del CE45.
Factor de crecimiento endotelial vascularSe ha observado que la expresión concomitante de metaloproteinasa 11 y del factor de crecimiento endotelial vascular (VEGF) está asociada con el riesgo de progresión de leucoplasias displásicas a CE46.
Características clínicasLa leucoplasia oral puede manifestarse clínicamente de diversas formas definidas a partir de: el patrón clínico (homogéneo o heterogéneo), la distribución o extensión de la lesión (focal o diseminada) y la localización dentro de la cavidad oral47.
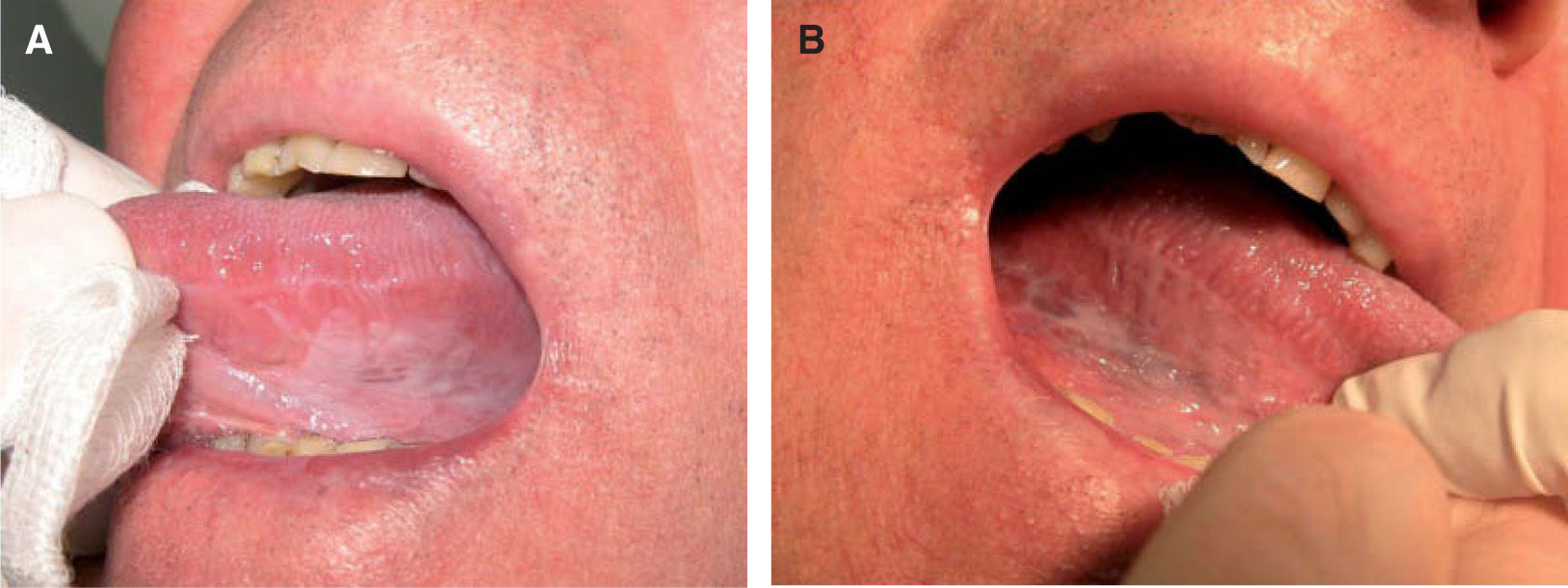
Patrón clínicoPatrón homogéneoDefinido como aquellas lesiones que presentan una superficie blanquecina regular y lisa con bordes bien delimitados48 (fig. 1). Este patrón clínico muestra un bajo riesgo de malignización a largo plazo (5 %)48-50.
Patrón heterogéneo
Incluye las leucoplasias que llevan asociado un componente eritematoso (eritroleucoplasia), nodular, erosivo, ulcerado o verrugoso exofítico. Su riesgo de malignización alcanza un 25 % de los casos, por lo que se consideran de alto riesgo15.
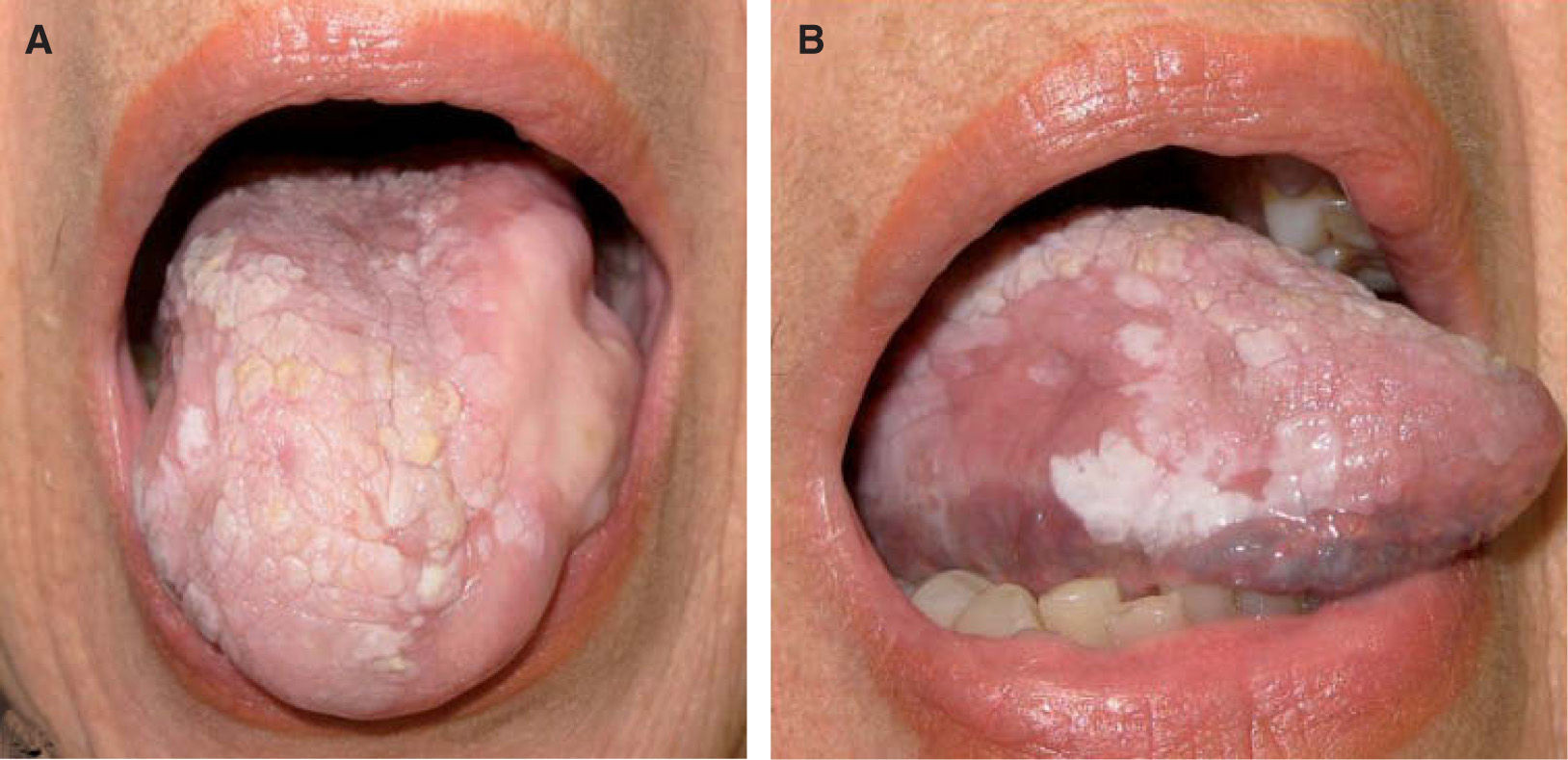
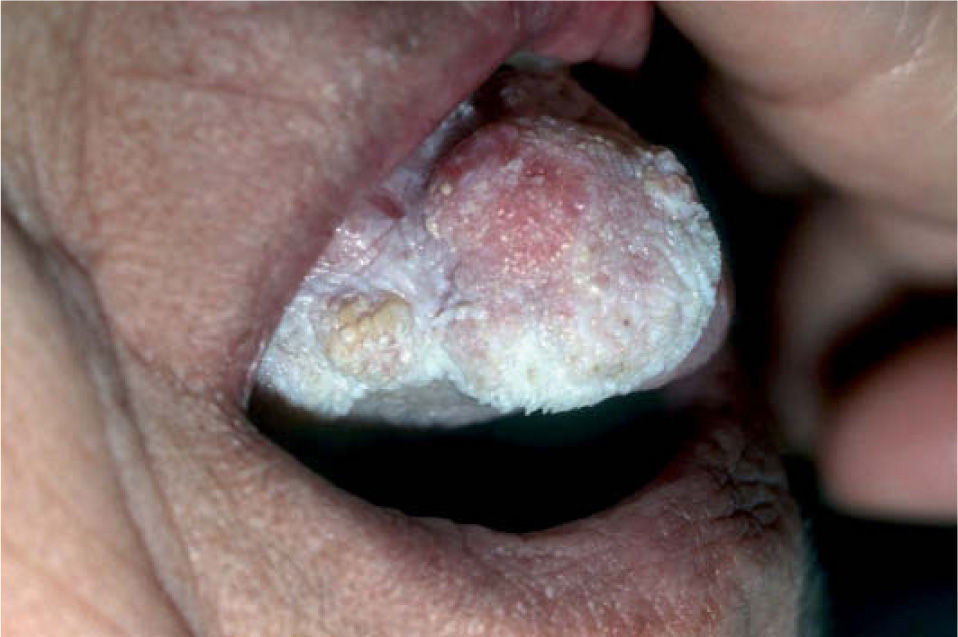
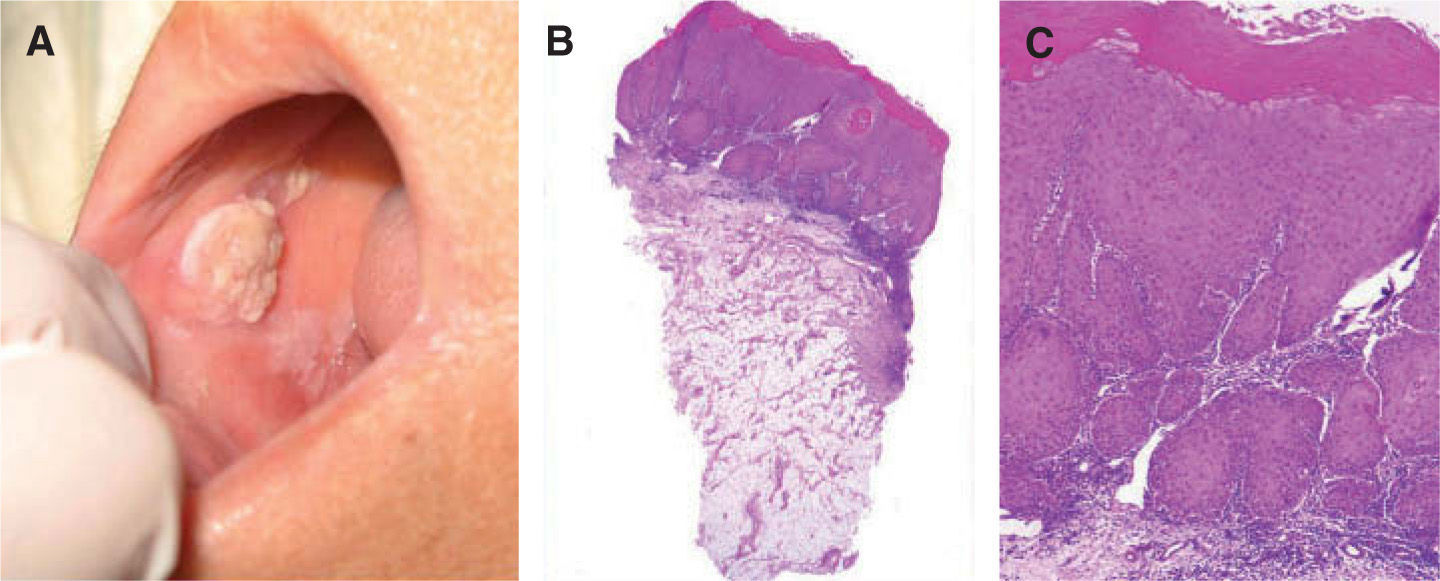
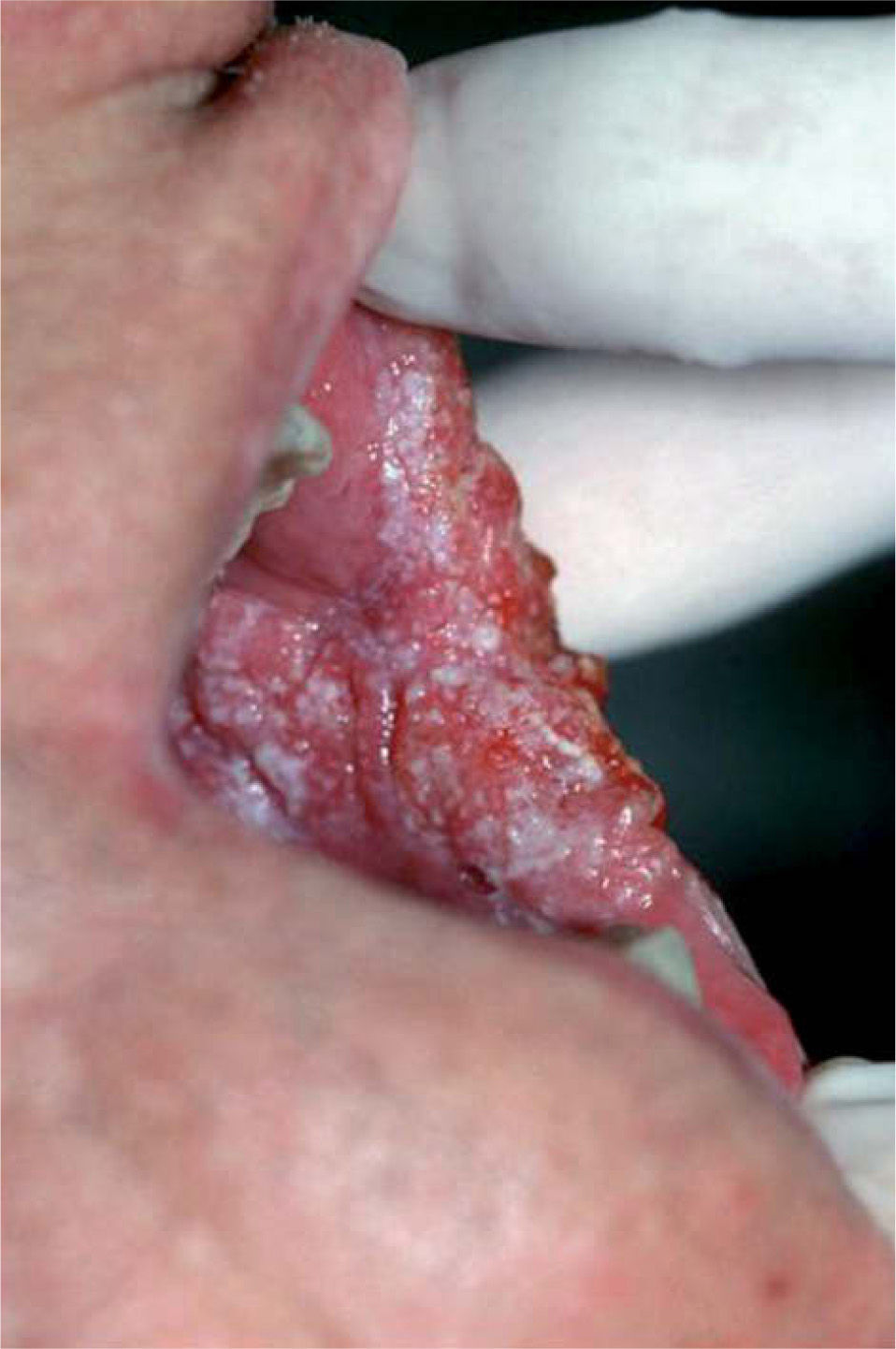
La leucoplasia verrugosa proliferativa (fig. 2), considerada actualmente como una entidad independiente del conjunto de leucoplasias, define un alto riesgo de malignización, alcanzando hasta el 80 % de los casos51,52. Se trata de una lesión que resulta difícil de distinguir clínicamente de un carcinoma verrugoso oral, también denominado papilomatosis oral florida o tumor de Ackerman (fig. 3). Ambas entidades aparecen en mujeres de edad avanzada, localizadas fundamentalmente en la región gingival, si bien progresivamente afectan a gran parte de la mucosa oral. Tienden a abarcar grandes áreas de mucosa con unos márgenes bien definidos. Se presentan clínicamente como lesiones blanquecinas verrugosas o como formas polipoides menos queratinizadas. El dato diferencial entre ambas entidades es histológico. En el carcinoma verrugoso, a diferencia de la leucoplasia verrugosa, aparece una marcada hiperplasia epitelial exo y endofítica que invade la dermis causando destrucción por compresión. La coexistencia de un CE o su desarrollo posterior, así como la displasia epidérmica, se detectó en el 63 % de una serie de 27 pacientes con leucoplasia verrugosa proliferativa por Slootweg y Müller 53, llegando a la conclusión de que esta variante de leucoplasia es un claro precursor de carcinoma.
Distribución de las lesiones

La distribución o extensión de la leucoplasia en la cavidad oral es un factor pronóstico de malignización a largo plazo. Así, la existencia de una leucoplasia focal se asocia a un buen pronóstico a largo plazo; por el contrario, las formas diseminadas, que afectan a varios puntos de la mucosa oral, suponen un peor pronóstico15.
Localización de las lesionesLas leucoplasias localizadas en el suelo de la boca y en la zona ventrolateral de la lengua se asocian a un mayor riesgo de malignización, con una tasa de transformación media del 43 %54. Ello se atribuye al hecho de que se trata de zonas más expuestas a carcinógenos presentes en la secreción salival, asociada a una mayor permeabilidad en dicho epitelio, tal y como indican estudios experimentales realizados en la mucosa oral55.
Otras característicasEl tamaño de la lesión mayor de 20mm, el rápido crecimiento de la leucoplasia, así como la historia previa de CE y el consumo habitual de alcohol o tabaco, conocidos carcinógenos de la mucosa orofaríngea, son otros factores de mal pronóstico a tener en cuenta en la evaluación de estos pacientes11.
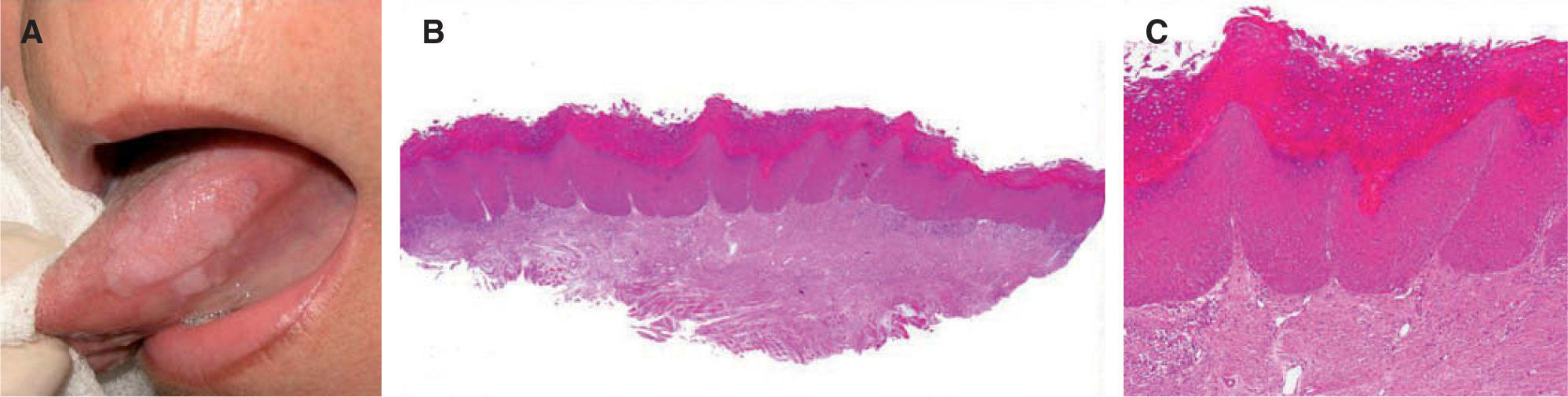
Características histopatológicasLa hiperqueratosis moderada y la hiperplasia epitelial no displásica son los hallazgos histológicos más frecuentemente descritos en la leucoplasia11,15 (fig. 4).
. C. Acantosis marcada en ausencia de cambios displásicos (hematoxilina-eosina, ×100).")
La displasia epitelial, caracterizada por un cuadro clínico inespecífico pero con datos histológicos bien definidos 56 (tabla 1), supone la expresión de un trastorno de maduración en el epitelio oral. Su presencia, que varía entre menos del 1 y el 30 %57, es globalmente aceptada como
uno de los factores predictores de malignidad58-61. Así, los pacientes que presentan leucoplasia oral con cambios displásicos desarrollan un CE hasta en un 36 % de los casos62 (fig. 5).
. C. Infiltrado neoplásico de células escamosas con marcada atipia celular y desestructuración arquitectural (hematoxilina-eosina, ×100).")
A. Carcinoma de células escamosas sobre leucoplasia heterogénea. B. Proliferación celular maligna que invade la dermis papilar y reticular superficial (hematoxilinaeosina, ×40). C. Infiltrado neoplásico de células escamosas con marcada atipia celular y desestructuración arquitectural (hematoxilina-eosina, ×100).
Actualmente el pronóstico y la actitud terapéutica frente a la leucoplasia se basa en el grado de displasia histológica, que incluye formas«leves», «moderadas»y«graves», dependiendo del espesor que ocupe el infiltrado atípico en el epitelio15. Esta clasificación tiene el inconveniente de la subjetividad interobservadora demostrada en diversos estudios63. Ésta es la causa por la cual resulta difícil establecer una buena correlación entre el grado de displasia y la tasa de transformación de la leucoplasia. Por otra parte, hay que tener presentes los siguientes matices: por un lado, la displasia epitelial no necesariamente evolucionará a CE en todos los casos, de hecho, en un discreto número de casos se ha observado incluso la resolución de la displasia histológica64-66. Por otro lado, la ausencia de la displasia histológica no excluye la posible malignización de una leucoplasia. Este dato se basa en el hecho de que esta variante sin displasia presenta una incidencia de transformación maligna del 15 %57,67. Con ello se concluye que la presencia de displasia histológica, independientemente de la gradación histológica, es considerada un importante factor predictor de la transformación neoplásica de la leucoplasia. No obstante, su presencia no es indispensable para que dicho fenómeno ocurra.
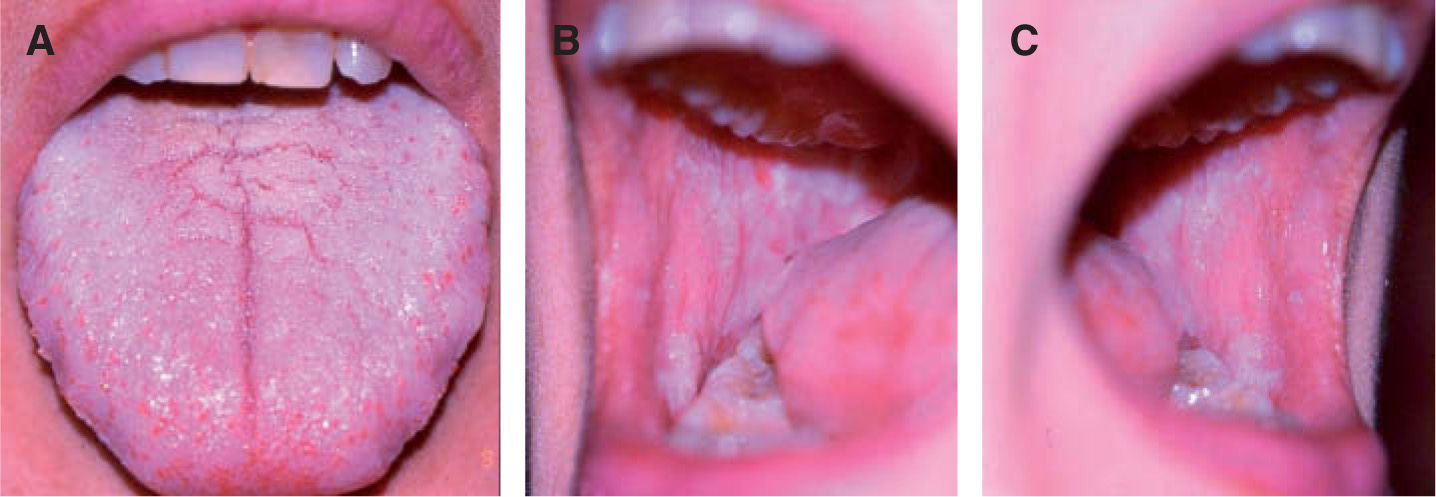
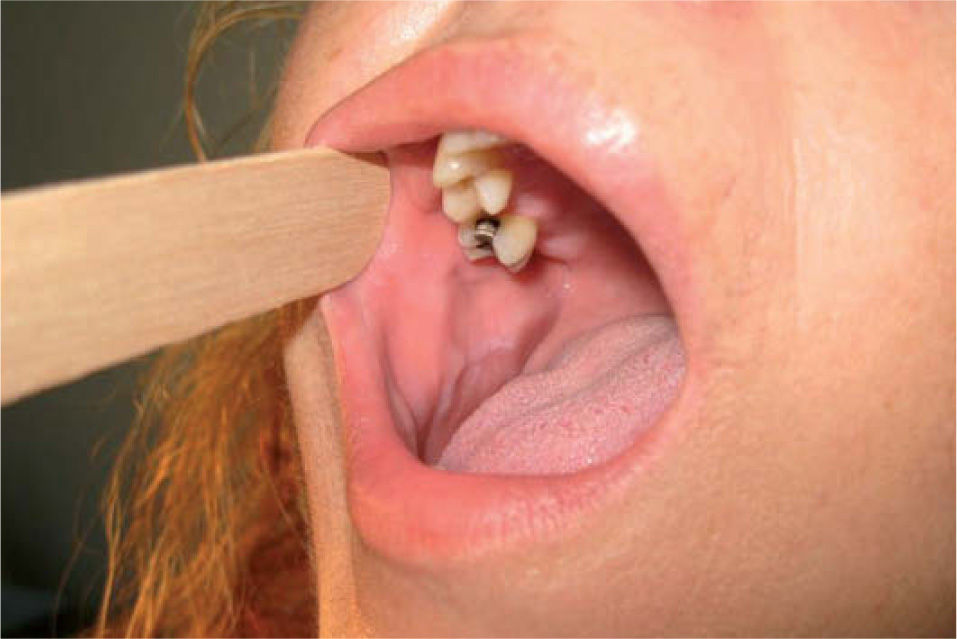
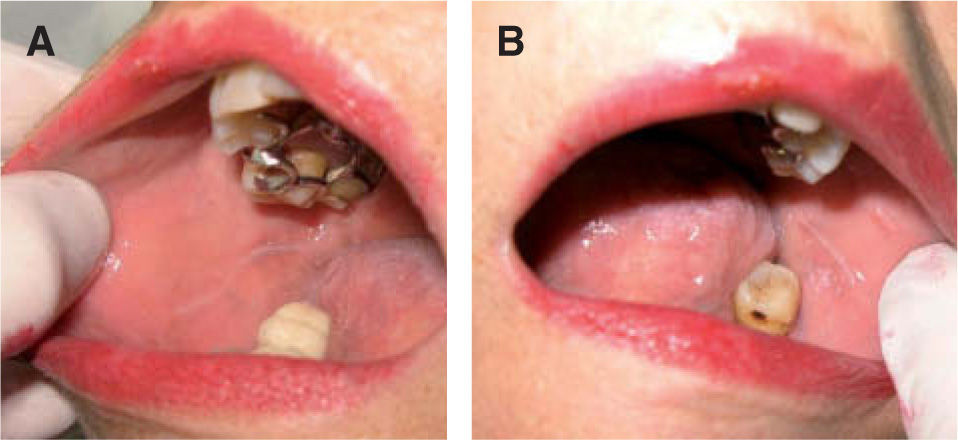
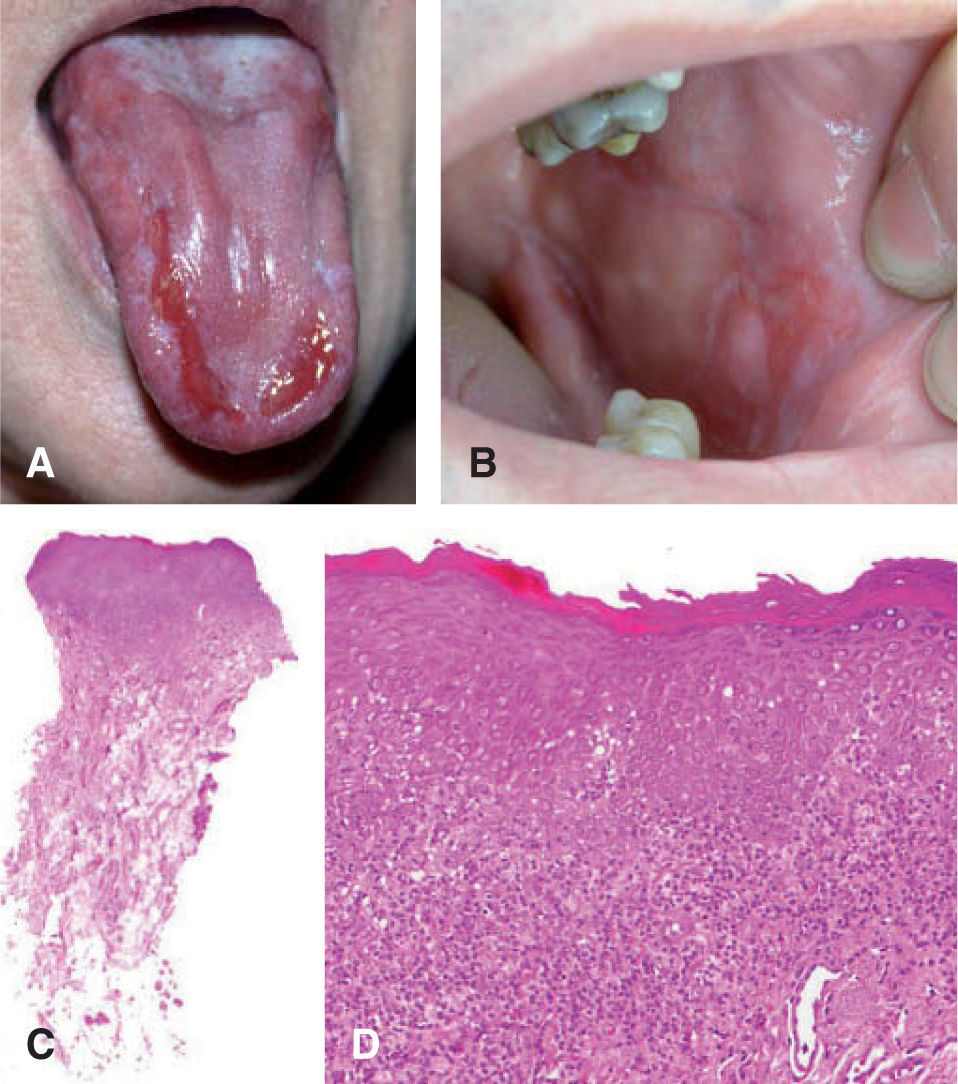
Diagnóstico diferencialEl diagnóstico de leucoplasia oral se establece después de descartar otras entidades muy bien definidas que pueden cursar con lesiones blanquecinas en la mucosa oral, como: leucoedema, nevo blanco espongiforme o«nevus de Cannon»(fig. 6), lesiones por mordedura o morsicatio buccarum (fig. 7), lesiones secundarias a dermatitis de contacto, estomatitis nicotínica o paladar del fumador, carcinoma escamoso (figs. 5 y 8), carcinoma verrugoso, leucoplasia oral vellosa, línea alba (fig. 9), lesiones orales del lupus eritematoso, liquen plano oral (fig. 10) y lúes secundaria. Las características clínicas más relevantes de estas entidades se incluyen en la tabla 211.
.")



. D. Infiltrado liquenoide compuesto mayoritariamente por linfocitos (hematoxilina-eosina, ×100).")
A y B. Liquen plano erosivo que afecta de forma simétrica al dorso de la lengua y a la mucosa yugal. C. Infiltrado inflamatorio que ocupa la dermis papilar y la dermis reticular superficial (hematoxilina-eosina, ×40). D. Infiltrado liquenoide compuesto mayoritariamente por linfocitos (hematoxilina-eosina, ×100).
Diferentes cuadros que cursan con lesiones blanquecinas en la mucosa oral
| Leucoedema | Lesión gris-blanquecina de la mucosa oral, benigna, que traduce una espongiosis intramucosa. Se localiza habitualmente en la mucosa de la mejilla y característicamente desaparece al estirar la superficie |
| Nevo blanco espongiforme; «nevus de Cannon»(fig. 6) | Genodermatosis autosómica dominante de expresividad variable que se caracteriza por la queratinización de la mucosa oral y, en ocasiones, también de la mucosa anal y vaginal. Se presenta como un engrosamiento esponjoso blanquecino que afecta de forma extensa al epitelio mucoso. No se ha observado la malignización de este proceso. Se presenta desde el nacimiento y la primera infancia, y puede asociarse a otras genodermatosis como la paquioniquia congénita o la disqueratosis congénita |
| Morsicatio buccarum o lesiones por mordedura (fig. 7) | Aspecto blanquecino y triturado de la mucosa bucal o labial en la línea de oclusión, causada por mordedura crónica. Las lesiones son benignas. El hábito es más frecuente en los individuos tensos o ansiosos |
| Lesiones por alergia de contacto | Placas blanquecinas en zonas de contacto con prótesis o empastes dentales, por sensibilización a diferentes agentes como la amalgama dental |
| Estomatitis nicotínica o paladar del fumador | Mucosa de color blanco grisáceo con pápulas umbilicadas correspondientes a las glándulas salivales localizadas en la mucosa palatina. Suele ser secundaria al hábito tabáquico con pipa. Resolución espontánea tras su cese |
| Carcinoma escamoso (figs. 5 y 8) | Placa blanquecina que histológicamente es una proliferación queratinocitaria pleomórfica que atraviesa la unión dermoepidérmica |
| Carcinoma verrugoso | Variante de carcinoma escamoso que se caracteriza por una superficie hipertrófica«en coliflor»e histológicamente por una invasión digitiforme o compresiva (bulldozering) |
| Leucoplasia oral vellosa | Cuadro que aparece en pacientes inmunodeprimidos asociado a la infección por el virus herpes tipo 8. Clínicamente se caracteriza por placas blanquecinas aterciopeladas que afectan de forma simétrica los bordes laterales de la lengua |
| Línea alba (fig. 9) | Cresta horizontal habitualmente hiperqueratósica localizada bilateralmente en la mucosa oral en la interdigitación de los dientes |
| Lupus eritematoso | Entre el 25 y el 40 % de los pacientes con lupus eritematoso presenta lesiones orales que afectan en particular al paladar y a la mucosa bucal. Las lesiones comienzan como parches rojos y evolucionan a erosiones irregulares y áreas de atrofia con o sin úlceras que, a menudo, curan dejando cicatriz |
| Liquen plano oral (fig. 10) | Placas dispuestas formando un retículo blanquecino característico, en ocasiones con ulceración secundaria, que ocupan habitualmente la mucosa yugal |
| Lúes secundaria | Placas de base ancha bien definida y nódulos planos con zonas erosivas asociadas. Se denominan condilomas planos, y también pueden estar presentes en la región anogenital |
La leucoplasia oral también se debe diferenciar de la queratosis benigna de la cresta alveolar (benign alveolar ridge keratosis), caracterizada por una placa blanquecina localizada en la región gingival de la cresta alveolar maxilar o mandibular, de probable origen traumático o friccional. Histológicamente muestra una marcada hiperortoqueratosis, así como una intensa hipergranulosis, con papilomatosis y acantosis con fusión de las crestas epidérmicas, sin infiltrado inflamatorio acompañante68,69.
TratamientoNo existe consenso en cuanto al tratamiento más apropiado para la leucoplasia oral. Dentro de las múltiples opciones terapéuticas (tabla 3), la eliminación de factores de riesgo (tabaco, alcohol) es una maniobra preventiva aplicable a todos los pacientes con leucoplasia56.
Opciones terapéuticas actuales
| Cese del tabaco/observación periódica |
| Escisión quirúrgica con o sin injerto |
| Criocirugía |
| Ablación con láser |
| Tratamiento antifúngico (leucoplasia asociada a Candida) |
| QuimioprevenciónRetinoidesVitaminas A, C y ECarotenosLicopeno |
| Terapia fotodinámica |
| Terapia tópicaBleomicinaVitamina A |
Dentro de la terapéutica no quirúrgica, una revisión de la base de datos Cochrane70 concluye que las intervenciones con bleomicina tópica, retinoides sistémicos y licopeno sistémico pueden ayudar a la resolución de la displasia, pero no existe suficiente evidencia de ello, dada la ausencia de estudios a largo plazo.
Dentro de las modalidades terapéuticas cruentas, las formas ablativas, que incluyen la criocirugía, el láser de CO2 y la resección quirúrgica, son las únicas opciones que presentan un nivel de evidencia aceptable para el control local de la leucoplasia a corto plazo71. Sin embargo, la alta tasa de fracaso terapéutico a largo plazo, la recurrencia local y el desarrollo de CE en el lugar de resección se justifican por varias razones:
La evidencia de una alteración molecular (pérdida de heterocigosidad) de los márgenes de resección histológicamente no afectos implica que en realidad no se alcanzan de manera habitual unos verdaderos márgenes libres de enfermedad24. Sudbo24 mostró en un estudio que el fenómeno de recurrencia y de malignidad se daba casi exclusivamente en aquellas leucoplasias con aneuploidia celular, en comparación con las leucoplasias con diploidia y tetraploidia. De esta manera, la forma leucoplásica aneuploide presentó un riesgo de transformación del 70 % a los 3 años de seguimiento, un alto riesgo de recidiva (el 30 % a los 3 años) y elevada letalidad (el 30 % en 3 años), a pesar de su resección completa, debido a la tendencia a afectar subclínicamente la mucosa circundante a la lesión. Así pues, la combinación del análisis histológico y molecular de los márgenes de resección de una leucoplasia permite establecer, con una mayor precisión, el riesgo de recidiva tumoral.
La naturaleza multifocal de la carcinogénesis oral hace que la extirpación quirúrgica local no sea un tratamiento definitivo en la erradicación del riesgo de desarrollo de carcinoma de cabeza y cuello4,72-74. Un último estudio demostró que el cáncer se desarrolló en el mismo lugar que la leucoplasia precedente en el 79 % de los casos. Desde el punto de vista de ploidia celular, el CE se localizó en el mismo lugar de la leucoplasia en el 100 % de las formas diploides, en el 81 % de las formas tetraploides y en el 73 % de las aneuploides27. Así pues, en los casos con aneuploidia, un porcentaje elevado de CE (27 %) se desarrolló en una localización diferente de la leucoplasia precedente, con una distancia media de 4,5cm (3–8,5cm).
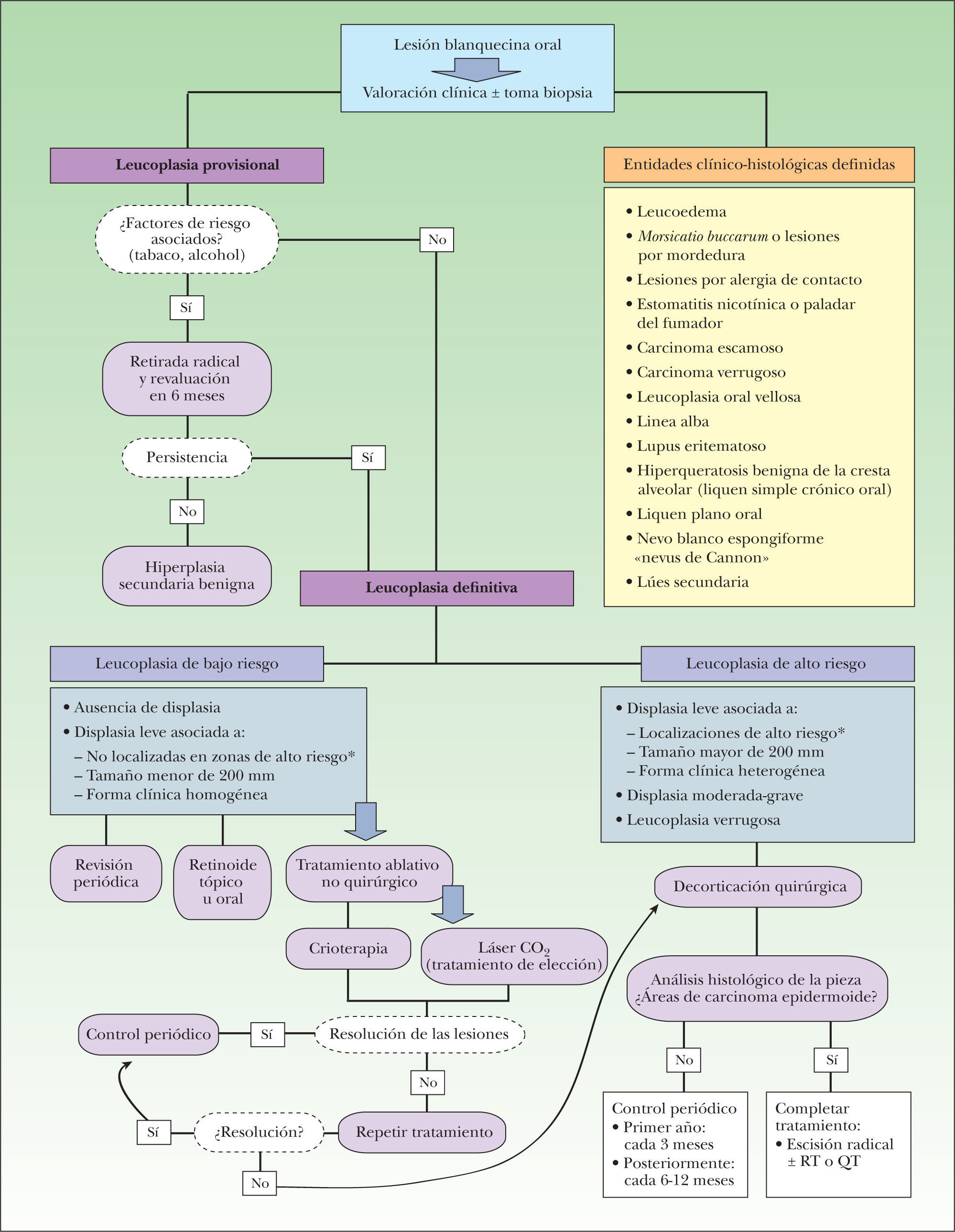
Por ello, el estudio de la leucoplasia mediante biología molecular nos permite predecir el riesgo de desarrollo de CE en el resto de la cavidad oral no afectada y establecer una pauta de seguimiento individual, según las características de cada paciente. En la actualidad, la actitud terapéutica (fig. 11) sigue teniendo como base la presencia o no de displasia histológica, que definiría dos grupos de riesgo:
- 1.
Grupo de bajo riesgo de malignización, formado por: a) aquellas leucoplasias con ausencia de displasia, y b) aquellas que presentan displasia leve y se localizan en áreas de bajo riesgo o muestran un espesor menor de 200mm o se presentan clínicamente como una leucoplasia homogénea.
En este grupo se podrían tomar diversas actitudes:
- –
Control periódico del paciente. El intervalo entre las visitas de seguimiento no debería superar los 12 meses con la finalidad de detectar cualquier cambio sugestivo de transformación maligna.
- –
Tratamiento de las lesiones con retinoides tópicos u orales. La experiencia en el uso de esta opción terapéutica muestra resultados bastante insatisfactorios, no consiguiendo la erradicación de las lesiones en la gran mayoría de los casos.
- –
Tratamiento mediante técnicas ablativas no quirúrgicas, tales como la crioterapia y la vaporización con láser de CO2. Dentro de estas opciones el láser ha mostrado mejores resultados en el control de las lesiones, por lo que se considera el tratamiento de elección en este grupo de bajo riesgo.
- –
- 2.
Grupo de alto riesgo de transformación maligna11, formado por: a) aquellas leucoplasias con displasia leve localizadas en zonas de alto riesgo o de más de 200mm de espesor o asociadas a una forma clínica heterogénea; b)las leucoplasias con displasia moderada o grave, y c)las leucoplasias verrugosas.

En este grupo está justificado el tratamiento agresivo quirúrgico consistente en la decorticación de todo el espesor de la mucosa en la que esté presente la leucoplasia.
Posteriormente se procede a un minucioso examen histológico de la pieza quirúrgica con el fin de descartar áreas de transformación neoplásica existente. En caso de detectar zonas de CE en la pieza extirpada se debería proceder a una escisión radical de la zona afectada asociada con otras terapias complementarias (radioterapia o quimioterapia).
El hecho de que la terapia actual no consiga un control óptimo de un gran porcentaje de leucoplasias establece la necesidad de desarrollar un tratamiento sistémico, que actúe de forma global sobre la mucosa oral: la quimioterapia oral.
La quimioprevención en Oncología oralLa quimioprevención es el uso de agentes farmacológicos o naturales que inhiben el desarrollo de cáncer invasivo. Dicha función la ejercen tanto por el bloqueo del daño del ADN que inicia la carcinogénesis, como por la apoptosis de las células premalignas en las cuales el daño celular ha ocurrido75. Las posibles dianas en la terapia molecular para la prevención del cáncer oral actualmente en fase de investigación incluyen la COX-2, el EGFR y el receptor de activación de proliferación de peroxisomas (PPAR gamma).
Inhibición de la ciclooxigenasa 2La COX-2 está sobreexpresada de forma selectiva en lesiones displásicas aneuploides, que son consideradas como las variantes con mayor potencial de malignidad76. Dicho fenómeno ocurre durante la transición maligna de la mucosa oral, que de alguna forma está relacionada con la presencia de inestabilidad genómica77.
Inhibición de receptor del factor de crecimiento epidérmicoLos inhibidores específicos de tirosín-quinasa para EGFR son dianas terapéuticas prometedoras en el control del carcinoma de cabeza y cuello78. El EGFR está sobreexpresado en la mayoría de las lesiones orales premalignas y malignas, y se asocia con un estadio avanzado y un descenso de la supervivencia79-81. HER2 (erbB-2), un miembro de la familia del EGFR, también se encuentra sobreexpresado en la carcinogénesis oral. De hecho, se ha observado que la terapia combinada de paclitaxel con PKI199 (inhibidor irreversible del dominio tirosín-quinasa del EGFR) prolonga la supervivencia en el cáncer de lengua mediante el incremento de la muerte celular programada82.
Terapias combinadas de inhibidores de COX-2/inhibidores de EGFRTorrance83 describió un mayor nivel de prevención de cáncer mediante el uso combinado de agentes contra dos dianas moleculares: COX-2 y EGFR. En concreto, EKB-569 (inhibidor irreversible del dominio tirosín-quinasa intercelular de EGFR) combinado con sulindaco (inhibidor COX no selectivo) demostró una mayor actividad en modelos animales de neoplasia intestinal.
En estudios de cultivos celulares procedentes de leucoplasias aneuploides se observó la existencia de una relación cruzada entre las vías de señalización de COX-2 y EGFR6. Ello establece que el bloqueo de ambas vías es necesario para una eficiente prevención del desarrollo del cáncer oral por su efecto sinérgico en el control de la leucoplasia. Este doble bloqueo también resultaría beneficioso en el control de los efectos nocivos producidos por el tabaco sobre la mucosa oral, ya que este agente oncogénico provoca la activación de EGFR que, secundariamente, genera un aumento de los niveles de COX-2 84. Por tanto, la inhibición combinada de EGFR y COX-2 es una estrategia prometedora en la prevención y en el tratamiento del cáncer de cabeza y cuello.
Fármacos agonistas de PPAR-gamma: antiinflamatorios no esteroideosEstudios epidemiológicos han demostrado la eficacia anticarcinogénica de los antiinflamatorios no esteroideos (AINE) en neoplasias fundamentalmente colorrectales, gástricas y esofágicas84-88. Su actividad antioncogénica está mediada por su efecto agonista sobre PPAR gamma. Ello puede inducir apoptosis a través de la activación de las cas- pasas89. Los AINE estudiados comprenden la indometacina, el ketoprofeno y el ibuprofeno, que son capaces de actuar como agonistas de PPAR gamma90.
Control y seguimientoDado el alto riesgo de recurrencia posquirúrgica y de desarrollo de CE de cabeza y cuello a distancia del foco leucoplásico primario, estos pacientes deben ser seguidos durante el resto de su vida a intervalos regulares que pueden variar entre 3–6 meses en aquellos pacientes considerados de alto riesgo, a 6–12 meses en aquellos pacientes considerados de bajo riesgo.
ConclusionesAnualmente se diagnostican en el mundo más de 300.000 casos de CE oral. Esta neoplasia epitelial agresiva conlleva una alta mortalidad y una importante morbilidad para aquellos pacientes que sobreviven, a pesar de los avances en el tratamiento mediante cirugía, radioterapia y quimioterapia. Su pronóstico sombrío no ha mejorado significativamente en las últimas 4 décadas. El fallo del tratamiento se basa en el desarrollo de segundos tumores primarios hasta en un 20 % de pacientes con CE en estadio precoz, y en la recurrencia local y metástasis en aquéllos localmente avanzados, que suponen dos tercios de los casos en el momento del diagnóstico. El tratamiento previo a este estado neoplásico puede reducir la incidencia anual de esta neoplasia agresiva.
Por ello, la leucoplasia, considerada la lesión precancerosa más frecuente en la población general, debe ser perfectamente caracterizada con la finalidad de definir aquella variante de«alto riesgo»con mayor potencial de transformación maligna. Los marcadores pronósticos tradicionales de las leucoplasias, como son las características clínicas y el grado de displasia epitelial oral, tienen un valor pronóstico limitado debido a la falta de reproducibilidad interobservador. El estudio rutinario en el futuro de diferentes parámetros de biología molecular, tales como la pérdida de heterocigosidad, la ploidia celular y la presencia de mutaciones en la p53, permitirá definir con mayor precisión aquellas leucoplasias de«alto riesgo», y con ello el establecimiento de una actitud terapéutica más agresiva frente a ellas.
Dentro de las diferentes opciones terapéuticas actuales, la incorporación futura del control histológico y molecular de los márgenes de exéresis, así como de terapias sistémicas que actúen contra diferentes dianas moleculares, permitirá un mejor control local y a distancia de la enfermedad.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.



