
The advent of biosimilar drugs and their use in routine clinical practice has revolutionized the management of moderate to severe psoriasis and led to changes in the positioning of the existing molecules used to control this condition. Evidence from clinical trials complemented by real-world experience has helped to clarify concepts and has significantly changed the use and positioning of biologic agents in this setting. The present document is an update on the position of the Spanish Psoriasis Working Group regarding the use of biosimilar drugs, which takes into account this new situation.
La incorporación de los fármacos biosimilares en el manejo de la psoriasis moderada-grave en nuestra práctica clínica diaria ha supuesto una revolución y un reposicionamiento de las diferentes moléculas que usamos diariamente para su control. La incorporación de evidencia mediante ensayos clínicos y experiencias de real world evidence ha ayudado a clarificar conceptos y modos de utilización que distan de su posicionamiento inicial. Es por ello por lo que se impone una actualización del posicionamiento del Grupo de Trabajo de Psoriasis en la utilización de los fármacos biosimiliares, dada la nueva realidad existente.
The first biosimilar medicines for use in dermatology in Spain were approved by the European Medicines Agency (EMA) in 2013.1 Almost ten years later, the purpose of this article is to provide an update on the position of the Spanish Psoriasis Group (GPs) on the use of biosimilars in dermatology—specifically the management of psoriasis—in our country and to present some analyses and reflections.
A biosimilar is a biological medicine highly similar to another biological medicine already approved in the European Union (EU), the so-called originator or reference drug. The two drugs are highly similar in terms of structure, biological function, efficacy, safety, and immunogenicity (the intrinsic ability of proteins and other biological medicinal products to provoke an immune response). The first biosimilar drug was approved by the European Commission in 2006.2 All biosimilars sold in Spain are subject to the European regulatory requirements governing these products. These regulations require the manufacturer to adequately demonstrate the comparability—mainly analytical, pharmacokinetic and pharmacodynamic—of each biosimilar with respect to its reference drug. Clinical trials for biosimilars usually focus on the most sensitive indication or indications of the originator and must include a sufficient number of patients to ensure statistical significance; the endpoint is usually non-inferiority. The findings of these studies are then extrapolated to other indications of the reference drug based on the prior demonstration of biosimilarity through comparability studies. The EMA is currently working on a pilot program aimed at advising manufacturers on how to reduce or eliminate the need for clinical trials in biosimilar development.3
Biosimilars must be approved by the EMA via a single European Union-wide procedure (the so-called centralized procedure). This involves a single application, which is evaluated by the Committee for Medicinal Products for Human Use (CHMP), a body in which representatives of the national agencies of the Member States participate. Once a favorable opinion has been issued by the CHMP, the European Commission is the body responsible for approving the drug, which is then authorized for sale throughout Europe. In Spain, a favorable financing resolution from the Ministry of Health is also required and the price of the product is set by the Interministerial Pricing Commission.4
Apart from the regulatory framework that has led to the availability of biosimilars for several TNFα inhibitors (etanercept, adalimumab and infliximab) in Spain, other important practical issues that have been evolving and maturing in the scientific community in recent years must also be considered. Concepts such as the extrapolation of indications (from a reference product to a biosimilar) under the regulatory framework and interchangeability (the non-medical substitution of a biosimilar for a reference drug) have generated uncertainty and very little pertinent data is available.4 On the other hand, the therapeutic arsenal for psoriasis has benefited from the addition of these cheaper biosimilar drugs which provide an opportunity to improve efficiency parameters within the health system and potentially to achieve significant savings in a complex therapeutic context. In recent years, this situation characterized by uncertainties and opportunities has led to a response in the form of new scientific evidence, clinical experience and the opinions of experts and scientific societies.
Registers and clinical practice studies in Spain and elsewhere have provided evidence on the effectiveness and safety of biosimilars in patients with psoriasis. We also have evidence, discussed below, to support switches between reference biologic agents and biosimilars. Furthermore, the inclusion (based on efficiency criteria) of biosimilars as an alternative first-line biologic agent in the treatment protocols of some Spanish autonomous communities has generalized their use among dermatologists in this country in recent years. This development has, to some degree, changed the therapeutic scenario for moderate to severe psoriasis, allowing earlier access to biologic drugs as a result of the application of slightly more lax severity criteria not so closely based on the Rule of Tens.
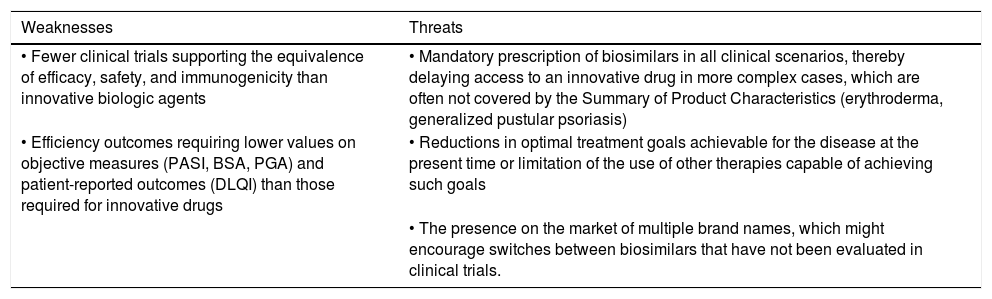
Table 1 summarizes the strengths, weaknesses, opportunities, and threats associated with the use of biosimilar drugs in the treatment of patients with moderate to severe psoriasis in Spain.
SWOT analysis of the use of biosimilars in moderate to severe psoriasis.
| Weaknesses | Threats |
|---|---|
| • Fewer clinical trials supporting the equivalence of efficacy, safety, and immunogenicity than innovative biologic agents | • Mandatory prescription of biosimilars in all clinical scenarios, thereby delaying access to an innovative drug in more complex cases, which are often not covered by the Summary of Product Characteristics (erythroderma, generalized pustular psoriasis) |
| • Efficiency outcomes requiring lower values on objective measures (PASI, BSA, PGA) and patient-reported outcomes (DLQI) than those required for innovative drugs | • Reductions in optimal treatment goals achievable for the disease at the present time or limitation of the use of other therapies capable of achieving such goals |
| • The presence on the market of multiple brand names, which might encourage switches between biosimilars that have not been evaluated in clinical trials. |
| Strengths | Opportunities |
|---|---|
| • Competitive pricing (sustainability of the SNS) | • To improve the access of patients with psoriasis to biologic therapy for disease control |
| • To standardize the situation across different autonomous communities, which currently have varying access guidelines and restrictions governing the use of biologic agents in the management of psoriasis. |
Abbreviations: BSA, body surface area; DLQI, Dermatology Life Quality Index; PASI, Psoriasis Area and Severity Index; PGA, Physician Global Assessment; SNS, Servicio Nacional de Salud.
In February 2013, the EMA approved the first biosimilar for use in immune-mediated diseases, which was based on infliximab; this was followed by a number of biosimilars for etanercept and adalimumab.5–7 Given patent expiration dates, biosimilars of certolizumab pegol, golimumab and ustekinumab are to be expected and are currently being developed by several companies; however, none of these products will be ready for submission to the EMA within the next 2 years.5 The pharmacokinetic and pharmacodynamic similarities between reference products and biosimilars are key to establishing biosimilarity. And we now have sufficient data from clinical trials to demonstrate that biosimilars are as effective and have the same drug survival prospects as their reference products.6,7
The increase in the number of biosimilars on the market in Spain is coupled with other considerations. While there is no national law regulating the use of biosimilars, we do have National Treatment Appraisal reports, which generally conclude with a section entitled “Final considerations of the Treatment Appraisal Coordination Group” specifying that “the selection of *** or another drug with high efficacy in this indication should take into account efficiency criteria”. However, in the case of tildrakizumab, guselkumab, and risankizumab the final considerations—for reasons that escape us and are not justified in the text—specify that these drugs can only be prescribed to “patients who have previously been treated with a biologic tumor necrosis factor (TNF) inhibitor” and, only in the case of guselkumab and risankizumab, “only when the use of a TNF inhibitor is contraindicated” (without specifying whether the TNF inhibitor must be a reference product or a biosimilar).8–10
Although data from the studies that led to the approval of several biosimilars can be reviewed in recent articles,5,11,12 in some cases the safety and efficacy data available are insufficient to support definitive conclusions, especially concerning switches between originator biologics and biosimilars and even between different biosimilars. In fact, higher rates of treatment discontinuation were observed in several studies that analyzed data following switches between originator biologic drugs and biosimilars.13 In some cases, discontinuation may be due to a nocebo effect, especially in diseases in which subjective parameters are used to assess therapeutic outcomes (rheumatic diseases) or when the patient has not been adequately informed about the biosimilar they are to receive. However, in other cases discontinuation may be due to differences in the composition of the excipient used or in the characteristics of the administration device, two elements that can lead to lower tolerability. For example, unlike originator adalimumab, all but one of the adalimumab biosimilars currently available in Spain have excipients that contain citrate, acetate, or lactate.
The EGALITY study, which was designed to demonstrate therapeutic equivalence between originator and biosimilar etanercept, did not analyze potential losses of efficacy when a patient was switched between the 2 products.14 In a study designed to assess the impact of multiple switches between an adalimumab biosimilar (Hyrimoz®/Halimatoz®/Hefiya®) and the reference product, the biosimilar demonstrated a good safety profile and equivalent retention rates.15 Likewise, no variations in the efficacy, safety or immunogenicity of the biosimilar were found in a recent meta-analyses of studies presenting data on switching from reference to biosimilar adalimumab in rheumatoid arthritis, psoriasis, or inflammatory bowel disease including a total of 2802 patients with different patterns of switching (including 6 studies in patients with psoriasis).12
Real-world evidence is just as important in the assessment of biosimilars as it is with innovative drugs. For example, data from the Spanish Registry of Systemic Treatments in Psoriasis (Biobadaderm) and the German Registry for Biological Treatment of Rheumatoid Arthritis (RABBIT) show that only 21% to 31% of patients on the registry would meet the criteria for inclusion in clinical trials.16 This means that evidence from registries and real-life clinical studies have greater external validity and applicability than clinical trial data in terms of what we might expect to encounter in clinical practice.
In the first article presenting data from the British Association of Dermatologist Biologic and Immunomodulators Register (BADBIR) on the use of adalimumab and etanercept biosimilars in patients with psoriasis, the biosimilars were shown to have safety and efficacy comparable to that of the originator biologics, although the sample size is small.17 In a larger sample, including 410 patients and a follow up of 549.84 person-years, Bellinato et al.8,10,18 report a one-year drug survival of 81.5% for biosimilar adalimumab, a finding indicating that its use as a first-line systemic treatment would increase the likelihood of drug survival, making this drug a cost-effective option in first-line biologic therapy.
According to recently published data on the use of biosimilars in 17 Spanish hospitals, the mean (SD) Psoriasis Area Severity Index (PASI) at the start of treatment with an adalimumab biosimilar was 7.4 (6.2),19 a value lower than that reported in the clinical trials. It should, however, be borne in mind that this sample included patients whose disease was adequately controlled with the reference drug prior to the switch and that treatment in real-world practice is started with lower PASI values than those required in clinical trials because physicians are not obliged to impose a clearance interval during which the patient receives no topical or systemic therapy. In that study, 70.8% of the patients had not received prior biologic therapy (biologic naïve) and 27% had concomitant psoriatic arthritis. It is interesting to note that the mean period during which the disease had been controlled with the originator drug before switching to the biosimilar in the 170 patients with prior treatment was 5.9 years and that the good response initially achieved with the reference drug was better retained in this subgroup than the response achieved in patients who started treatment with biosimilar adalimumab de novo, in other words, without prior therapy with the reference drug.19
Published data on the use of an infliximab biosimilar and on the switch between the reference product and the biosimilar CT-P13 in 165 Japanese patients show that drug survival was lower for the biosimilar when it was used as a second-line therapy.20,21 The treatment-response outcome used in the Japanese study was an absolute PASI value of less than 1, close to the desired outcome in real-world clinical practice,9 and better response rates were observed in male patients and patients with arthritis.21
Finally, a narrative review by Strand et al.22 summarizing immunogenicity data for biosimilars in patients with rheumatic diseases, plaque psoriasis or inflammatory bowel disease found that biosimilars had immunogenicity parameters similar to those of the reference products. In psoriasis, the evidence has been more consistent with adalimumab and etanercept, while in the case of infliximab, a drug used almost exclusively in patients with inflammatory bowel disease, immunogenicity has not been directly investigated.14,15,23
The findings cited above should be considered together with the evidence from studies on large cohorts of patients with psoriasis, which suggests that early treatment of plaque psoriasis with biologic agents as opposed to phototherapy or topical treatments alone may decrease the incidence of psoriatic arthritis.24,25
Position Papers of Other Spanish Scientific SocietiesThe positions of other Spanish scientific societies on the use of biosimilars in various immune-mediated diseases are of interest to the position of the GPs on biosimilars, although many of them may be outdated in light of more recent developments following the incorporation of biosimilar drugs into clinical practice in Spain.
Spanish Association of Biotechnology Companies (AseBIO) (2008)The position of AseBIO26 regarding the extrapolation of indications is that it is acceptable provided that a high level of biosimilarity is demonstrated between the biosimilar and the reference product in terms of analytical data—including chemical structure, manufacturing process and quality controls—and of preclinical and clinical data. They further stipulate that when an indication is extrapolated (with due justification) that it must be complemented with adequate post-authorization commitments. They also stipulate that the Summary of Product Characteristics must specify which data relate specifically to the biosimilar product and which are extrapolated from the reference drug, in addition to other elements that must form an integral part of this public reference document. They stress that pharmacy and therapeutics committees in hospitals must rigorously evaluate all these aspects when considering the use of biosimilars, especially when contemplating an automatic switch from a reference drug to a biosimilar product in patients already receiving treatment.
Another point made in this position statement is that, given our inability to predict the presence and severity of immunogenicity and its variability across different populations, this risk should be clearly established in each population before allowing the extrapolation of indications.
Finally, they also highlight the absence of scientific evidence to support the possibility of safely exchanging one biologic product for another bearing the same international nonproprietary name when the 2 products are produced by different manufacturers.
Spanish Society of Medical Oncology (SEOM) (2018)The position of our oncologist colleagues in the SEOM27 is that studies are needed to provide follow-up data on the classic parameters that can demonstrate unequivocal benefit—such as drug survival and progression-free drug survival—to facilitate the assessment of comparability in the extrapolation process. Moreover, particular attention must be paid to ensuring that the mechanism of action of the biosimilar is mediated by the same receptors and mechanisms as that of the reference product since the mode of action of some substances is complex and often involves multiple receptors and sites of action, which could be important in the extrapolation of indications.
In general, SEOM advises against the substitution of one biologic for another for the same indication in cancer patients once treatment has begun because of the lack of evidence and the risk that patients who experience adverse effects or progressions following a change in medication may attribute these events to the switch. Since the course of neoplastic diseases is subject to frequent changes it is important not to give patients any cause for confusion or mistrust relating to the factors that might have influenced the course of their disease: it is important that such changes should not be erroneously attributed to a switch from one medication to another, especially if the switch was made without the express consent of both the prescribing physician and the patient.
Proper pharmacovigilance measures require that the clinician and the patient know at all times what drug the patient is taking and that the drug prescribed has not been substituted as a result of supply problems or for any other reason without the express consent of the prescribing physician and the patient.
Spanish Society of Rheumatology (SER) (2018)In their position papers,28,29 the SER considers it essential to preserve the right of physicians to prescribe a specific drug which has been selected taking into account the individual characteristics and circumstances of each patient without, of course, ignoring the economic impact of such decisions.
They further specify that the decision to switch a patient from an originator biologic to a biosimilar should be made only by the prescribing physician and with the patient's consent. When the patient's condition is stable, switching from a reference drug to a biosimilar may be acceptable, but switches must be decided on a case-by-case basis and with the patient's consent.
The SER also considers that the relevant hospital authorities must ensure that all biologic and biosimilar drugs funded by the Spanish health system for the management of rheumatic diseases should be available in all the hospitals in the National Health System.
Traceability is a key element in the quality control of biologic drugs because it makes it possible to assign suspected adverse reactions to a specific batch and product. When the reference biologic drug has more than one indication, the extrapolation of indications for the biosimilar must conform to EMA standards.
The optimal use of biosimilars requires continuous dialog and interaction between physicians, hospital pharmacies, patient associations, and regulatory bodies to preserve the patients’ right to health and to offer them high quality, safe and effective medications.
Spanish Society of Ocular Inflammation (SEIOC) (2019)SEIOC's position agrees with those of the other scientific societies and stresses the fact that continuous dialog and interaction between physicians and patient associations is key to a more widespread understanding and acceptance of biosimilars as high quality, safe and effective medicines.30
Spanish Society of Hospital Pharmacists (SEFH) (2015)The SEFH position offers the most forward-looking vision regarding the introduction of biosimilar medicines into the current therapeutic arsenal.31 The following is a summary.
- 1.
Biosimilars are regulated by a robust EMA framework.
- 2.
Biosimilars are safe medicines.
- 3.
Indications can be extrapolated if biosimilarity and comparable safety and efficacy between the reference drug and the biosimilar have been demonstrated for one indication.
- 4.
Interchangeability between reference drugs and biosimilars is increasingly supported by better evidence.
- 5.
Hospital pharmacy and therapeutics committees and regional committees play a key role in the evaluation of biosimilars and their inclusion in hospital protocols. These committees establish the criteria for using biosimilars, including protocols for treatment switching and monitoring, as they do for all other biologic drugs.
- 6.
The traceability of biosimilars must be guaranteed.
- 7.
Information on biosimilar drugs must be made available.
- 8.
The use of biosimilars favors a more sustainable healthcare system.
- 9.
Hospital pharmacists play a key role in the pharmaco-therapeutic management of biosimilars.
In Spain, the national government defines the criteria and procedures for setting the prices of drugs and medical devices funded by the Spanish national health system (Sistema Nacional de Salud [SNS]).32 Since the introduction of biosimilars, their use has been one of the most important determinants of total spending on drugs. According to the latest SNS data, biologic drugs account for half of total hospital spending on medicines.33 This has led to the implementation of measures aimed at encouraging the use of biosimilar drugs in hospitals, although their penetration across different Spanish autonomous communities is uneven.
It is important to note that the main benefit of the advent of biosimilars is improved access to biologic therapy for patients who have few therapeutic alternatives and for whom they are indicated, an aspect that has been studied and applied in several real-world clinical practice series. However, the presence on the market of reference drugs with biosimilars should not limit the use of innovative drugs without biosimilars in patients with more severe or complex conditions.9
The need to ensure patient acceptance, the main aim of all treatment, requires that medical, nursing and pharmacy professionals should all be well informed so that they can resolve patient doubts and limit any inconvenience these treatments may entail. Training programs are needed to resolve the doubts of professionals involved in the use of biosimilars and to draw up joint “shared benefit” strategies involving management, pharmacy personnel and the clinical professionals involved in the efficient management of biologic therapies.
Conclusions and GPs Position- •
A biosimilar is a biologic medicine containing a version of an active substance already approved as a biotechnological drug (the reference medicine) in the European Economic Area, for which the patent rights have expired. All biosimilars must conform to the strict regulatory requirements laid down by the EMA.
- •
When the active substance is a protein, the amino acid sequence must be the same as that of the reference drug and the biosimilar must meet all the criteria for biosimilarity in terms of structure (analytical) and biological and therapeutic activity (equivalence studies).
- •
Biosimilarity must be demonstrated in accordance with the EMA regulatory framework, which has been under development since 2004. This includes the following requirements: comparative studies of quality; studies of non-clinical parameters (pharmacodynamics, effects on physiological and cellular targets); clinical studies (excluding studies on safety and efficacy, which have already been demonstrated for the reference drug); pharmacokinetic and pharmacodynamic studies. If applicable, clinical trials demonstrating equivalence of efficacy, safety, immunogenicity are also required and, in some cases, additional data supporting the extrapolation of indications.
- •
The dosage and route of administration of the biosimilar must be the same as those of the reference product.
- •
Any divergence in terms of potency, pharmaceutical form, excipients, or presentation must be properly justified, and safety can never be compromised. Changes associated with an improvement in the efficacy of the biosimilar as compared to the reference product are incompatible with the authorization of a biosimilar. However, changes made to improve the safety of the treatment (such as reductions in impurities or lower immunogenicity) should be reported but do not preclude biosimilarity.
- •
The extrapolation of indications to those of the reference biologic shall be based on the data available for each biosimilar, and is a process regulated by the EMA.
- •
Interchangeability refers to the possibility of replacing one drug with another without the patient experiencing any changes in clinical effect. Before September 2022, decisions on the interchangeability of biosimilars were made by national health authorities of Member States, but the EMA and the Heads of Medicines Agencies now consider that once a biosimilar product has been approved for use in the EU it is considered to be interchangeable with its reference product and with other biosimilars of the same reference product. By contrast, the FDA only authorizes switches between the reference drug and the biosimilar and vice versa, but not between biosimilars of the same reference product.
- •
Treatment switches should be decided by the prescribing physician and/or according to protocols agreed by the hospital's Pharmacy and Therapeutics Committee. All switches must be accepted and monitored by the prescribing clinician and the patient must always be informed.
- •
The decision to allow automatic substitution (a switch from the originator biologic to a biosimilar implemented by the pharmacist without the intervention of the prescribing clinician) is the responsibility of each country and such substitions are not permitted in Spain.
- •
Since biosimilars are comparable to the reference drug in efficacy, safety and immunogenicity, the focus of their added value is on reducing cost and improving efficiency. It is important, however, that efficiency be calculated using objective cost-effectiveness studies that take into account each situation. For example, in the case of an early switch following the first-line use of a biosimilar due to a lack of efficacy or safety, the efficiency of the biosimilar should be compared to the cost of treatment with the innovative drugs introduced as second-line therapy. The GPs considers efficiency studies carried out using appropriate methodology to be opportune and essential.
- •
The availability of efficient biosimilars in the effectiveness/safety balance should favor a paradigm shift that would allow them to become first-line therapies in the management of moderate to severe psoriasis requiring systemic treatment.
- •
The availability of biosimilars should not lead physicians to reduce the optimal treatment goals achievable for the disease at the present time or to limit the use of other therapies that could achieve such goals.
- •
The use of biosimilars as first-line therapies is appropriate in a high percentage of patients with moderate to severe psoriasis requiring biologic therapy, based mainly on efficiency criteria. However, this positioning must also be considered in the context of evidence that there may be other biologic drugs available that can provide better efficacy and safety, as demonstrated by the results of comparative clinical trials and meta-analyses. In specific cases, therefore, the option of using a first-line biologic drug for which no biosimilar is available should be available to physicians and patients.
- •
The correct introduction and management of biosimilar drugs in each hospital requires multidisciplinary coordination involving the resource management department, hospital pharmacists, clinicians, clinical pharmacology, and nursing services. In Spain, unlike other EU countries, biologic drugs are classified as medications that can only be dispensed by a hospital pharmacy for outpatient use (MHDA), which means that their prescription and dispensing is restricted to the hospital setting.
- •
The benefits derived from the rational use of resources through the introduction of biosimilars should have a positive effect on the health care circuit that can be achieved through shared benefit strategies.
- •
Finally, it would be advisable to standardize the situation across the autonomous communities in Spain. Currently the different health systems serving the autonomous communities have different access guidelines and restrictions governing the use of biologics in the management of psoriasis.
- •
For reasons of efficiency, it is advisable to start biologic therapy with biosimilars in most patients. If a contraindication exists or if treatment must be discontinued due to adverse effects or an inadequate response, it should be possible for physicians, at their own discretion, to prescribe the most appropriate treatment (taking into account efficiency criteria) irrespective of its mechanism of action or pharmacological target.
R. Ruiz-Villaverde has served as principal investigator or sub-investigator (PI/SI) and/or received honoraria as a speaker, advisory board member, and/or consultant for Abbvie, Celgene, Leo Pharma, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, UCB, Pfizer, and MSD.
M. Galán-Gutiérrez has served as a PI/SI and/or received honoraria as a speaker, advisory board member, and/or consultant for Abbvie, Celgene, Leo Pharma, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, UCB, Pfizer, and MSD.
Mar Llamas-Velasco has served as a PI/SI and/or received honoraria as a speaker, advisory board member, and/or consultant for Abbvie, Celgene, Leo Pharma, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, UCB, Pfizer, and MSD.
Laura Salgado-Boquete has served as a PI/SI and/or received honoraria as a speaker, advisory board member, and/or consultant for Abbvie, Celgene, Leo Pharma, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, UCB, Pfizer, and MSD.
Lluis Puig has served as a PI/SI and/or received honoraria as a speaker, advisory board member, and/or consultant for Abbvie, Almirall, Amgen, Baxalta, Biogen, Boehringer Ingelheim, Celgene, Gebro, Janssen, JS BIOCAD, Leo-Pharma, Lilly, Merck-Serono, MSD, Mylan, Novartis, Pfizer, Regeneron, Roche, Sandoz, Samsung-Bioepis, Sanofi, and UCB.
Pablo de la Cueva has served as an investigator, consultant, and/or speaker for the following pharmaceutical companies: Abbvie, Almirall, Amgen, Astellas, Biogen, BMS, Boehringer, Celgene, Gebro, Janssen, LEO Pharma, Lilly, MSD, Novartis, Pfizer, Sandoz, Sanofi, and UCB.
José Manuel Carrascosa has served as a PI/SI and/or received honoraria as a speaker, advisory board or steering committee member for Abbvie, Novartis, Janssen, Lilly, Sandoz, Amgen, Almirall, BMS, Boehringer ingelheim, Biogen, and UCB.
M. Teresa Abalde Pintos, Ignacio Alonso García, María Luisa Alonso Pacheco, Alsina Gibert Mercé, Gloria Aparicio Español, Mariano Ara Martín, Susana Armesto Alonso, Antoni Azón Masoliver, Ferrán Ballescá López, Ofelia Baniandres Rodríguez, Didac Barco Nebreda, Álvaro Barranquero Fernández, Ana Batalla Cebey, Isabel Bielsa Marsol, Xavier Bordas Orpinell, Leopoldo Borrego Hernando, Rafael Botella Estrada, Jesús María Careaga Alzaga, Rafael Carmena Ramón, Gregorio Carretero Hernández, Ana María Carrizosa Esquivel, José Manuel Casanova Seuma, Alberto Conde Taboada, Marisol Contreras Stelys, Pablo Coto Segura, Esteban Daudén Tello, Carlos de la Torre Fraga, Rubén del Río Gil, Aleisandre Docampo Simón, Noemí Eiris Salvado, Juan Escalas Taberner, Esther Eusebio Murillo, José Manuel Fernández Armenteros, Emilia Fernández López, M. Luisa Fernández Díaz, Almudena Fernández Orland, Carlos Ferrándiz Foraster, Marta Ferrán Farrés, Lara Ferrándiz Pulido, Eduardo Fonseca Capdevila, Manuel Galán Gutiérrez, Francisco Javier García Latasa de Araníbar, Ptableilar García Muret, Vicente García-Patos Briones, Marta García Bustínduy, Ignacio García Doval, Rosa García Felipe, Alicia L. González Quesada, Beatriz González Sixto, Alfonso González Morán, Teresa Gárate Ayastui, Francisco José Gómez García, José Manuel Hernanz Hermosa, M. Isabel Hernández García, Pedro Herranz Pinto, Enrique Herrera Ceballos, Marta Herrera Sánchez, Rafael Jesús Jiménez Puya, Enrique Jorquera Barquero, Rosario de Fátima Lafuente Urrez, Salvador V. Laguarda Porter, Mónica Larrea García, M. del Mar Llamas Velasco, Anna López Ferrer, Jesús Luelmo Aguilar, Pablo Lázaro Ochaita, José Luis López Estebaranz, María Marcellán Fernández, Amparo Marquina Vila, Eugenio Marrón Moya Servando, Trinidad Martín González, Antonio Martorell Calatayud, Francisco Javier Mataix Díaz, Almudena Mateu Puchades, Carolina Medina Gil, M. Victoria Mendiola Fernández, Miren Josune Michelena Eceiza, Jordi Mollet Sánchez, José Carlos Moreno Giménez, Carlos Muñoz Santos, Antoni Nadal Nadal, Belén Navajas Pinedo, Jaime Notario Rosa, Francisco Peral Rubio, Narciso Pérez Oliva, Celia Posada García, Josep A. Pujol Montcusi, Conrado Pujol Marco, Silvia Pérez Barrio, Amparo Pérez Ferriols, Beatriz Pérez Suárez, Trinidad Repiso Montero, Miquel Ribera Pibernat, Raquel Rivera Díaz, Vicente Rocamora Durán, Jesús Rodero Garrido, Sabela Rodríguez Blanco, M. del Carmen Rodríguez Cerdeira, Lourdes Rodríguez Fernández-Freire, Manuel Ángel Rodríguez Prieto, Jorge Romani de Gabriel, Alberto Romero Maté, Mónica Roncero Riesco, Cristina Rubio Flores, José Carlos Ruiz Carrascosa, Diana Patricia Ruiz Genao, Ricardo Ruiz Villaverde, Montserrat Salleras Redonnet, Jorge Santos-Juanes Jiménez, María José Seoane Pose, Patricia Serrano Grau, Estrella Simal Gil, Caridad Soria Martínez, José Luis Sánchez Carazo, Manuel Sánchez Regaña, M. Dolores Sánchez-Aguilar Rojas, Rosa Taberner Ferrer, Lucía Tomás Aragonés, Francisco Valverde Blanco, Ricardo Valverde Garrido, Francisco Vanaclocha Sebastián, Manel Velasco Pastor, Diana Velázquez Tarjuelo, Asunción Vicente Villa, David Vidal Sarró, Jaime Vilar Alejo, Eva Vilarrasa Rull, Marta Vilavella Riu, Rosario Vives Nadal, Hugo Alberto Vázquez Veiga, Juan Ignacio Yanguas Bayona, and Ander Zulaica Gárate.
The members of the Appraisal Committee of the Spanish Psoriasis Group (GPs) are listed in Appendix 1.


