






















Ichthyoses are a heterogeneous group of diseases sharing symptoms and a common etiopathogenic mechanism. Clinically, these diseases are characterized by the presence of erythema and variable degrees of skin thickening and desquamation. Although the affected area, severity, and molecular substrate are very variable, they are all signs of a disruption of the barrier formed during epidermal differentiation. Ichthyoses follow patterns of Mendelian inheritance and present symptoms since birth or shortly thereafter. Clinically, they can be categorized into non-syndromic (when symptoms are caused exclusively by the epidermal barrier dysfunction) and syndromic ichthyoses (when the causal gene has extracutaneous functions impacting other organs).
Knowledge of molecular mechanisms has improved dramatically over the past few years, and we currently know not only most causal genes, but also the functions of the encoded proteins and their impact on skin barrier formation. In the first part of this review, we’ll be introducing ichthyosis physiopathology, along with a clinical and genetic update of non-syndromic entities (those included in the consensus classification and those clinically and/or molecularly characterized since then).
Las ictiosis son un grupo heterogéneo de enfermedades que comparten síntomas y un mismo mecanismo etiopatogénico. Desde el punto de vista clínico, estas enfermedades se caracterizan por la presencia de eritema y diferentes grados de engrosamiento y descamación cutáneos. Aunque el área afectada, la gravedad y el sustrato molecular son muy variables, todas ellas representan la manifestación de una disrupción de la barrera que se forma durante el proceso de diferenciación epidérmica. Las ictiosis siguen patrones de herencia mendeliana y causan síntomas desde el nacimiento o poco tiempo después. Desde el punto de vista clínico, se dividen en ictiosis no sindrómicas (cuando los síntomas están causados únicamente por el defecto de la barrera epidérmica) e ictiosis sindrómicas (cuando el gen causal también tiene funciones extracutáneas que determinan manifestaciones en otros órganos).
El conocimiento de las bases moleculares ha avanzado extraordinariamente en los últimos años y conocemos no solo la mayoría de los genes que las ocasionan, sino la función de las proteínas que codifican y el impacto en la formación de la barrera cutánea. En la primera parte de este trabajo hacemos una introducción a la fisiopatología de las ictiosis, así como una actualización clínica y genética de las entidades no sindrómicas, tanto las incluidas en la última clasificación consenso como otras que se han caracterizado clínica o molecularmente a lo largo de los últimos años.

Ichthyoses are a heterogeneous group of diseases that share symptoms and the same etiopathogenic mechanism. Clinically these diseases are characterized by the presence of erythema and different degrees of cutaneous thickening and desquamation. Although the affected area, severity, and molecular substrate are highly variable, they all represent the manifestation of a disruption of the barrier formed during the process of epidermal differentiation. Therefore, this group of diseases is grouped under the name disorders of epidermal differentiation (DED).1
Epidermis is a tissue with a very well-demarcated structure: the constantly dividing stem cells responsible for regenerating the epithelium (keratinocytes) are located in the stratum basale and the cells generated from this division undergo a gradual process of differentiation as they migrate toward the surface.2 This differentiation process (called cornification) causes morphological changes in keratinocytes due to changes in protein expression patterns, reorganization of the cytoskeleton, lipid secretion, and establishment of intercellular junctions, culminating in the formation of the stratum corneum, which is the most superficial layer of the epidermis, in which keratinocytes have terminally differentiated into corneocytes, cells lacking nuclei and organelles whose cytoplasm is completely filled with structural proteins. Corneocytes form junctions between them called corneodesmosomes and are embedded in a lipid matrix secreted in the lower layers that saturates the intercellular space and in which enzymes capable of degrading the intercellular junctions are immersed3 (Fig. 1).

Structure of the epidermis. Stem cells in the stratum basale differentiate to form the stratum corneum, composed of corneocytes packed with structural proteins, anchored by intercellular junctions, and embedded in an extracellular lipid matrix.
The cornification process generates an organic structure that acts as a barrier between the environment and the rest of the organism. Structural proteins give cells mechanical resistance to physical and chemical aggressions; corneodesmosomes bind the cells together and transfer this mechanical resistance to the entire tissue; lipids seal the intercellular spaces, preventing water exchange and, therefore, both the entry of pathogens and water lost through evaporation; and finally, enzymes secreted into the intercellular space are responsible for the orderly degradation of intercellular junctions, allowing the epithelium to desquamate and making it harder for parasites to colonize the skin. In addition, the thinness of the stratum corneum makes the epidermal barrier a very flexible structure, capable of carrying out its functions without limiting the movement of the organism.4
An alteration of the epidermal barrier that prevents it from carrying out its functions exposes the organism to physical, chemical, and biological aggressions leading to transepidermal water loss. As a homeostatic response, the body increases the production of differentiated cells and thickens the stratum corneum, which leads to the appearance of the common sign of DED, hyperkeratosis and increased desquamation. Thickening of the stratum corneum causes skin elasticity loss, which hinders mobility and can lead to fissure formation. Transepidermal water loss increases the risk of dehydration and, along with the hypohidrosis associated with these disorders, heat intolerance. In addition, the barrier defect increases immune system exposure to pathogens, causing hyperactivation of inflammatory pathways, erythema, and pruritus.5,6
When this alteration of the epidermal barrier is caused by extrinsic factors, the disease is called acquired ichthyosis. It is a finding associated with certain tumors (Hodgkin's disease, multiple myeloma and cutaneous T-cell lymphoma), endocrine (renal failure, diabetes, hyperparathyroidism), autoimmune (lupus erythematosus and dermatomyositis), and infectious diseases (leprosy, mycobacterial, or HIV infections), and nutritional disorders (usually lipid or vitamin metabolic alterations), or certain drugs (cholesterol-lowering drugs, allopurinol, acitretin, EGFR and BRAF inhibitors). Acquired ichthyosis is a late onset disease—years or decades after birth—its symptoms are usually mild vs all the other DED, and its treatment is usually directed against the extrinsic factor causing the disease.7
Inherited DED respond to Mendelian inheritance patterns and cause symptoms since birth or a few months later. At present, more than 100 genes have been described with pathogenic variants causing some type of DED. The phenotypic presentation of these diseases is very heterogeneous and depends on the functions of the affected protein. Table 1 shows the role of the genes involved in the appearance of ichthyoses in epidermal differentiation.1 Consequently, clinical diagnosis can be difficult, and definitive confirmation of the disease requires molecular confirmation.
Function of the main genes involved in congenital ichthyosis pathogenesis.
| Gene (MIM code) | Function |
|---|---|
| Structural proteins | |
| Keratins | |
| KRT1 (139350) | Contributes to the intermediate filament cell cytoskeleton in suprabasal epidermal cells |
| KRT2 (600194) | Contributes to the intermediate filament cell cytoskeleton in the more superficial suprabasal epidermal cells |
| KRT10 (148080) | Contributes to the intermediate filament cell cytoskeleton in suprabasal epidermal cells, less important in palmoplantar skin |
| Filaggrin | |
| ASPRV1 (611765) | Protease involved in filaggrin processing due to phenotype it probably targets additional proteins |
| CASP14 (605848) | Protease involved in filaggrin processing |
| FLG (135940) | Protein that aggregates keratin intermediate filaments through promotion of disulfide-bond formation and liquid-liquid phase separation. It is part of the cornified cell envelope and upon proteolysis contributes to the formation of the epidermal natural moisturizing factor |
| POMP (613386) | Molecular chaperone responsible for promoting proteasome formation and thereby for the maturation of proteins critical for epidermal differentiation such as filaggrin |
| Cornified envelope | |
| LORICRIN (152445) | Precursor protein of the cornified envelope |
| TGM5 (603805) | Enzyme that crosslinks precursor proteins of and to the cornified envelope |
| Lipid metabolism | |
| Ceramides | |
| ABCA12 (607800) | Enzyme involved in ceramide loading to the lamellar bodies |
| ABHD5 (604780) | Functions as an acyltransferase and as a coactivator of adipocyte triglyceride lipase |
| ALDH3A2 (609523) | Enzyme involved in fatty acid synthesis, which some studies suggest are used as ceramide precursors |
| ALOX12B (603741) | Enzyme involved in ceramide crosslinking to the cornified envelope to form the corneocyte lipid envelope |
| ALOXE3 (607206) | Enzyme involved in ceramide crosslinking to the cornified envelope to form the corneocyte lipid envelope |
| CERS3 (615276) | Synthesizes ceramide from modified ultra-long chain fatty acid and dihydrosphingosine |
| CYP4F22 (611495) | ω-Hydroxylation of ultra-long chain fatty acids for acylceramide synthesis |
| ELOVL1 (611813) | Fatty acid elongase involved in ultra-long chain fatty acid synthesis, which are ceramide precursors |
| ELOVL4 (605512) | Fatty acid elongase involved in ultra-long chain fatty acid synthesis, which are ceramide precursors |
| GBA1 (606463) | Catalyzes the breakdown of the glycolipid glucosylceramide to ceramide and glucose |
| KDSR (136440) | Synthesizes ceramide precursor dihydrosphingosine from serine |
| LIPN (613924) | Unclear role in ceramide synthesis |
| NIPAL4 (609383) | Unclear role in ceramide synthesis |
| PEX7 (601757) | Plays an essential role in peroxisomal protein import (including phytanoyl-coenzyme A hydroxylase) |
| PHGDH (606879) | Enzyme involved in serine synthesis, which is used as a ceramide precursor |
| PHYH (602026) | Enzyme involved in synthesis of peroxisomal fatty acids, which are used as ceramide precursors |
| PNPLA1 (612121) | Catalyzes ω-O-esterification with linoleic acid to form acylceramides |
| PSAT1 (610936) | Enzyme involved in serine synthesis, which is used as a ceramide precursor |
| PSPH (172480) | Enzyme involved in serine synthesis, which is used as a ceramide precursor |
| SDR9C7 (609769) | Enzyme involved in ceramide crosslinking to the cornified envelope to form the corneocyte lipid envelope |
| SLC27A4 (604194) | Adds coenzyme A to ultra-long chain fatty acids for ceramide synthesis |
| TGM1 (190195) | Enzyme with poorly understood functions in ceramide crosslinking to the cornified envelope to form the corneocyte lipid envelope |
| UGCG (602874) | Glycosylates acyl-ceramide |
| Cholesterol | |
| EBP (300205) | Enzyme involved in cholesterol synthesis |
| MBTPS2 (300294) | Membrane metalloprotease involved in activation of transcription factors involved in cholesterol enzyme transcription |
| NSDHL (300275) | Enzyme involved in cholesterol synthesis |
| SREBF1 (184756) | Transcription factor involved in cholesterol enzyme transcription |
| STS (300747) | Synthesizes cholesterol from cholesterol sulfate |
| SULT2B1 (604125) | Responsible for cholesterol sulfation, which plays a major role in the regulation of epidermal differentiation |
| SUMF1 (607939) | Responsible for modifying various sulfatases |
| Lamellar bodies | |
| SNAP29 (604202) | Mediates lamellar body fusion events |
| VIPAS39 (613401) | Mediates lamellar body fusion events |
| VPS33B (608552) | Mediates lamellar body fusion events |
| Dolichol | |
| DOLK (610746) | Phosphates dolichol |
| MPDU1 (604041) | Adds mannose to dolichols as preparation for protein O-glycosylation and N-mannosylation |
| PIGL (605947) | Involved in glycosylphosphatidylinositol anchor synthesis |
| SRD5A3 (611715) | Involved in dolichol synthesis |
| Intercellular junctions | |
| Tight junctions | |
| CLDN1 (603718) | Tight junction protein, controls paracellular permeability |
| CLDN10 (617579) | Tight junction protein, controls paracellular permeability |
| Gap junctions | |
| GJA1 (121014) | Gap junction protein, controls intercellular communication |
| GJB2 (220290) | Gap junction protein, controls intercellular communication |
| GJB3 (603324) | Gap junction protein, controls intercellular communication |
| GJB4 (605425) | Gap junction protein, controls intercellular communication |
| GJB6 (604418) | Gap junction protein, controls intercellular communication |
| Desmosomes | |
| CDSN (602593) | Component of corneodesmosomes in the stratum corneum, increases mechanical resistance |
| DSG1 (125670) | Desmosome protein, ensures intercellular adhesion |
| DSP (125647) | Desmosome protein, ensures intercellular adhesion |
| FLG2 (616284) | Ensures cell-cell adhesion in the upper epidermal layers in a corneodesmosin-dependent fashion |
| Proteases e inhibitors | |
| SPINK5 (605010) | Serine protease inhibitor, prevents junction degradation |
| CAST (114090) | Cysteine protease inhibitor, prevents junction degradation |
| CSTA (184600) | Cysteine protease inhibitor, prevents junction degradation |
| SERPINB8 (601697) | Serine protease inhibitor, prevents junction degradation |
| ST14 (606797) | Functions as an epithelial membrane activator for other proteases and plays a role in profilaggrin processing and hair follicle growth |
| Transcription/translation | |
| AARS1 (601065) | Alanyl-tRNA synthetase |
| ERCC2 (126340) | Component of the TFIIH complex involved in nucleotide excision repair and type 2 gene transcription |
| ERCC3 (133510) | Component of the TFIIH complex involved in nucleotide excision repair and type 2 gene transcription |
| GTF2E2 (189964) | Component of the TFIIE complex involved in type 2 gene transcription |
| GTF2H5 (608780) | Component of the TFIIH complex involved in nucleotide excision repair and type 2 gene transcription |
| MARS1 (156560) | Methionyl-tRNA synthetase |
| RNF113A (300951) | Ring finger protein involved in pre-mRNA splicing |
| TARS1 (187790) | Threonyl-tRNA synthetase |
| Calcium channels | |
| TRPM4 (606936) | Calcium activated-ion channel, associated with proliferation regulation |
| Clathrin-coated vesicles | |
| AP1B1 (600157) | Part of the clathrin-coated vesicle adaptor complex |
| AP1S1 (603531) | Part of the clathrin-coated vesicle adaptor complex |
| Miscellaneous | |
| MPLKIP (609188) | Interacts with cyclin dependent and polo kinases, maintains cell cycle integrity |
MIM: gene code in OMIM database.
According to the consensus classification of congenital ichthyoses published in 2010,8 congenital ichthyoses are categorized into non-syndromic (when all symptoms are caused by the epidermal barrier defect and there are only cutaneous signs) and syndromic ichthyoses (when the causal gene has extracutaneous functions that determine manifestations in other organs).9 Following this criterion, we have included diseases described later or that have been better characterized in recent years, updating them both clinically and molecularly.
Non-syndromic ichthyosesThese are ichthyoses whose genetic alterations affect only the differentiation of the epidermis. They are subcategorized based on the time of onset, inheritance pattern, phenotypic spectrum, or type of genetic involvement (Table 2).8
Non-syndromic ichthyosis classification.
| Non-syndromic ichthyoses |
|---|
| Ichthyosis vulgaris (IV) (ORPHA: -) |
| Recessive X-linked ichthyosis (RXLI) (ORPHA: 461) |
| Autosomal recessive congenital ichthyosis (ARCI) (ORPHA: 281097) |
| Lamellar ichthyosis (ORPHA: 313) |
| Congenital ichthyosiform erythroderma (ORPHA: 79394) |
| Harlequin ichthyosis (ORPHA: 457) |
| Self-healing collodion baby (ORPHA: 281122) |
| Acral self-healing collodion baby (ORPHA: 281127) |
| Bathing suit ichthyosis (ORPHA: 100976) |
| Keratinopathic ichthyoses (ORPHA: 281103) |
| Autosomal dominant epidermolytic ichthyosis (ORPHA: 312) |
| Autosomal recessive epidermolytic ichthyosis (ORPHA: 512103) |
| Annular epidermolytic ichthyosis (ORPHA: 281139) |
| Ichthyosis Curth-Macklin (ORPHA: 79503) |
| Ichthyosis with confetti (ORPHA: 281190) |
| Superficial epidermolytic ichthyosis (ORPHA: 455) |
| Epidermolytic nevus (ORPHA: 497737) |
| Peeling skin syndromes (ORPHA: 817) |
| Generalized peeling skin syndrome (ORPHA: 263543) |
| Acral peeling skin syndrome (ORPHA: 263534) |
| PLACK Syndrome (ORPHA: 44138) |
| Erythrokeratoderma (ORPHA: 79355) |
| Loricrin keratoderma (ORPHA: 79395) |
| KLICK syndrome (ORPHA: 281201) |
KLICK: keratosis linearis, ichthyosis congenita, and sclerosing keratoderma (queratosis linear, ictiosis congénita y queratoderma esclerosante); ORPHA: código de la enfermedad en la base de datos ORPHANET; PLACK: peeling skin, leukonychia, acral punctate keratosis, cheilitis, and knuckle pads (piel descamada, leuconiquia, keratosis puntada acral, queilitis y almohadillas en los nudillos).
It is the most common form of ichthyosis in northern European populations, with a prevalence of 1 in 80 in English patient cohorts.10 IV is characterized, in most cases, by a relatively late presentation, a few months after birth. Patients exhibit a fine, whitish scaling, and, although it is most prominent on the extensor surface of the extremities, it is not uncommon for the entire body surface to be affected. Occasionally, patients have larger, lighter brown scales on the extensor areas of the extremities, especially the lower ones. Another characteristic feature of IV is palmar hyperlinearity, a finding known as filaggrin palm11 (Fig. 2). Palmoplantar keratoderma is a much less common finding.12 Additionally, many patients suffer from itching, and scratching leads to very expressive whitish linear lesions. Although IV is frequently associated with atopic dermatitis, only half of the patients with this inflammatory disease associate pathogenic variants in FLG.13 IV is caused by semi-dominant pathogenic variants in FLG14—a gene encoding the filaggrin protein—the major protein inside corneocytes. Of note, there are 2 more rarer forms of ichthyosis that also affect filaggrin metabolism, caused by recessive pathogenic variants in CASP1415 and dominant pathogenic variants in ASPRV1.16 While the first form exhibits a phenotype similar to IV, the second one, exceptionally, exhibits a phenotype similar to lamellar ichthyosis—see below—but with palmar hyperlinearity and no collodion membrane at birth.16
Recessive X-linked ichthyosis (RXLI) in a patient with a pathogenic variant in FLG.")
RXLI affects only men (with a prevalence of 1 in 5000)17 as it is caused by deletions of the steroid sulfatase gene (STS), located in a distal region of the short arm of the X chromosome that does not undergo inactivation.18 Although women are carriers of the disease and transmit it, they only manifest it in the rare cases in which the gene deletion occurs in both chromosomes.19 RXLI is characterized by the presence of dark brown polygonal scales on the extensor surfaces of the extremities (Fig. 3). Although the size of the scales is variable, they tend to be larger on the extensor surfaces of the lower extremities. Involvement of the flexures is variable and probably depends on the severity of desquamation being present in the most clinically expressive subjects. Desquamation affects the scalp, neck, and retroauricular region, giving a false appearance of insufficient hygiene to some of these patients. Palms and soles are usually spared. As with IV, many patients with RXLI are born with a normal skin and manifest the disease after the first few months of life, yet they still some have some sort of presentation at birth similar to yet much milder than a collodion membrane. The deficit in STS function prevents desulfation of cholesterol sulfate in the epidermis, hindering adequate desquamation of the horny layer. It also prevents desulfation of dehydroepiandrosterone sulfate, hindering cervical maturation during labor, causing prolonged second stage of labor in carrier women, which can help confirm clinical suspicion.20 Although RXLI is considered a non-syndromic ichthyosis, 30%-40% of patients present with attention deficit hyperactivity disorder, so it is very likely that some steroid metabolized by the STS enzyme plays a role as a neurotransmitter.21

IV and RXLI may have a similar appearance in the more symptomatic IV cases. Family history screening—involvement of maternal grandfather or mother's siblings—normal appearance of palms and soles and sparing of flexures in most patients with RXLI may help differentiate the 2 entities. The confluence of pathogenic variants in FLG and STS in some patients may significantly increase the expressiveness of desquamation in RXLI patients and facilitate association of palmoplantar hyperlinearity.22
Autosomal recessive congenital ichthyosis (ARCI)ARCI alludes to its inheritance pattern, has a very low prevalence, estimated to be between 7.2 and 16.2 affected per 106 inhabitants both in Spain23 and other regions.24,25 Unlike ichthyoses in the previous group, ARCIs debut at birth. The most typical form of presentation is called collodion baby, which is characterized by a shiny and smooth transparent glue-like membrane that covers the entire body and, in most severe cases, causes ectropion (eversion of the eyelids) and eclabium (eversion of the lips). The collodion membrane is shed within the first few weeks of life, progressing into a phenotypic presentation of variable severity, with varying degrees of erythema and different desquamation morphology. Accordingly, 5 different clinical forms are distinguished and detailed below. In cases where no collodion membrane is seen, neonates present with erythroderma and varying degrees of hyperkeratosis and desquamation. In most cases, the affected genes are involved in the metabolism of ceramides,8 a lipid component of the intercellular matrix that prevents transepidermal water loss (Fig. 4) (Table 3). Although there is no exact geno-phenotypic correlation, patients with pathogenic variants in TGM1 associate alopecia, ectropion, and neonatal presentation as a collodion baby with a significantly higher frequency than the other genes.8
 and a fatty acid (green) to form acylceramide, which can be released as a free lipid (yellow) or bound to proteins (orange). Mutations in the genes depicted cause non-syndromic (black) or syndromic (red) ichthyosis.")
Ceramide synthesis pathway in the epidermis. These reactions involve the union of a sphingoid base (blue) and a fatty acid (green) to form acylceramide, which can be released as a free lipid (yellow) or bound to proteins (orange). Mutations in the genes depicted cause non-syndromic (black) or syndromic (red) ichthyosis.
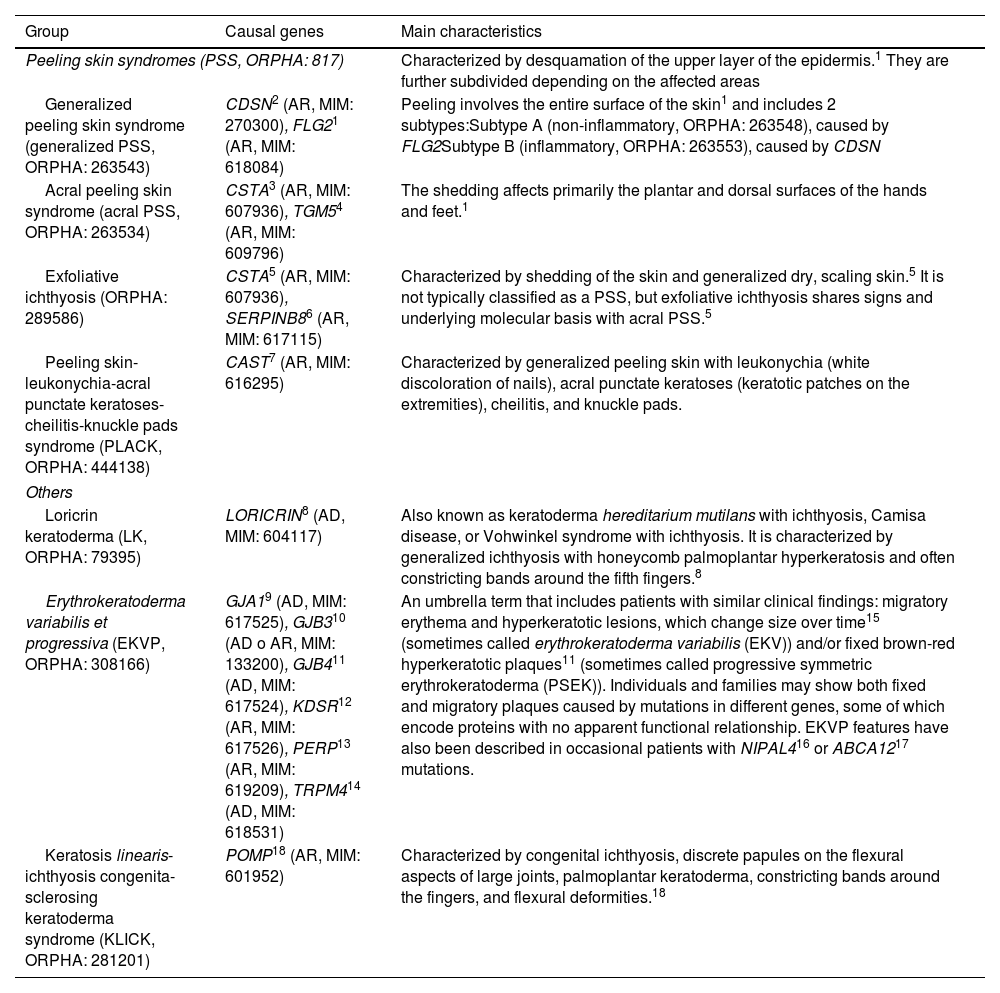
Rare forms of non-syndromic ichthyoses.
| Group | Causal genes | Main characteristics |
|---|---|---|
| Peeling skin syndromes (PSS, ORPHA: 817) | Characterized by desquamation of the upper layer of the epidermis.1 They are further subdivided depending on the affected areas | |
| Generalized peeling skin syndrome (generalized PSS, ORPHA: 263543) | CDSN2 (AR, MIM: 270300), FLG21 (AR, MIM: 618084) | Peeling involves the entire surface of the skin1 and includes 2 subtypes:Subtype A (non-inflammatory, ORPHA: 263548), caused by FLG2Subtype B (inflammatory, ORPHA: 263553), caused by CDSN |
| Acral peeling skin syndrome (acral PSS, ORPHA: 263534) | CSTA3 (AR, MIM: 607936), TGM54 (AR, MIM: 609796) | The shedding affects primarily the plantar and dorsal surfaces of the hands and feet.1 |
| Exfoliative ichthyosis (ORPHA: 289586) | CSTA5 (AR, MIM: 607936), SERPINB86 (AR, MIM: 617115) | Characterized by shedding of the skin and generalized dry, scaling skin.5 It is not typically classified as a PSS, but exfoliative ichthyosis shares signs and underlying molecular basis with acral PSS.5 |
| Peeling skin-leukonychia-acral punctate keratoses-cheilitis-knuckle pads syndrome (PLACK, ORPHA: 444138) | CAST7 (AR, MIM: 616295) | Characterized by generalized peeling skin with leukonychia (white discoloration of nails), acral punctate keratoses (keratotic patches on the extremities), cheilitis, and knuckle pads. |
| Others | ||
| Loricrin keratoderma (LK, ORPHA: 79395) | LORICRIN8 (AD, MIM: 604117) | Also known as keratoderma hereditarium mutilans with ichthyosis, Camisa disease, or Vohwinkel syndrome with ichthyosis. It is characterized by generalized ichthyosis with honeycomb palmoplantar hyperkeratosis and often constricting bands around the fifth fingers.8 |
| Erythrokeratoderma variabilis et progressiva (EKVP, ORPHA: 308166) | GJA19 (AD, MIM: 617525), GJB310 (AD o AR, MIM: 133200), GJB411 (AD, MIM: 617524), KDSR12 (AR, MIM: 617526), PERP13 (AR, MIM: 619209), TRPM414 (AD, MIM: 618531) | An umbrella term that includes patients with similar clinical findings: migratory erythema and hyperkeratotic lesions, which change size over time15 (sometimes called erythrokeratoderma variabilis (EKV)) and/or fixed brown-red hyperkeratotic plaques11 (sometimes called progressive symmetric erythrokeratoderma (PSEK)). Individuals and families may show both fixed and migratory plaques caused by mutations in different genes, some of which encode proteins with no apparent functional relationship. EKVP features have also been described in occasional patients with NIPAL416 or ABCA1217 mutations. |
| Keratosis linearis-ichthyosis congenita-sclerosing keratoderma syndrome (KLICK, ORPHA: 281201) | POMP18 (AR, MIM: 601952) | Characterized by congenital ichthyosis, discrete papules on the flexural aspects of large joints, palmoplantar keratoderma, constricting bands around the fingers, and flexural deformities.18 |
AD: autosomal dominant, AR: autosomal recessive.
1. Alfares A, Al-Khenaizan S, Al Mutairi F, Fuad C, Mutairi A, Abdullah K. Peeling skin syndrome associated with novel variant in FLG2 gene. Am J Med Genet A. 2017;173;3201–3204. https://doi.org/10.1002/AJMG.A.38468.
22. Oji V, Eckl KM, Aufenvenne K, Nätebus M, Tarinski T, Ackermann K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet. 2010;87;274–281. https://doi.org/10.1016/J.AJHG.2010.07.005.
3. Muttardi K, Nitoiu D, Kelsell DP, O’Toole EA, Batta K. Acral peeling skin syndrome associated with a novel CSTA gene mutation. Clin Exp Dermatol. 2016;41;394–398. https://doi.org/10.1111/CED.12777.
4. Cassidy AJ, van Steensel MAM, Steijlen PM, van Geel M, van der Velden J, Morley SM, et al. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am J Hum Genet. 2005;77;909–917. https://doi.org/10.1086/497707.
5. Blaydon DC, Nitoiu D, Eckl KM, Cabral RM, Bland P, Hausser I, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell–cell adhesion. Am J Hum Genet. 2011;89;564–571. https://doi.org/10.1016/J.AJHG.2011.09.001.
6. Pigors M, Sarig O, Heinz L, Plagnol V, Fischer J, Mohamad J, et al. Loss-of-function mutations in serpinb8 linked to exfoliative ichthyosis with impaired mechanical stability of intercellular adhesions. Am J Hum Genet. 2016;99;430–436. https://doi.org/10.1016/J.AJHG.2016.06.004.
7. Lin Z, Zhao J, Nitoiu D, Scott CA, Plagnol V, Smith FJD, et al. Loss-of-function mutations in CAST cause peeling skin, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads. Am J Hum Genet. 2015;96;440–447. https://doi.org/10.1016/J.AJHG.2014.12.026.
8. Maestrini E, Monaco AP, McGrath JA, Ishida-Yamamoto A, Camisa C, Hovnanian A, et al. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel's syndrome. Nat Genet. 1996;13;70–77. https://doi.org/10.1038/ng0596-70.
9. Boyden LM, Craiglow BG, Zhou J, Hu R, Loring EC, Morel KD, et al. Dominant de novo mutations in GJA1 cause erythrokeratodermia variabilis et progressiva, without features of oculodentodigital dysplasia. J Invest Dermatol. 2015;135;1540–1547. https://doi.org/10.1038/JID.2014.485.
10. Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet. 1998;20;366–369. https://doi.org/10.1038/3840.
11. Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, et al. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet. 2000;67;1296–1301. https://doi.org/10.1016/S0002-9297(07)62957-7.
12. Boyden LM, Vincent NG, Zhou J, Hu R, Craiglow BG, Bayliss SJ, et al. Mutations in KDSR cause recessive progressive symmetric erythrokeratoderma. Am J Hum Genet. 2017;100;978–984. https://doi.org/10.1016/J.AJHG.2017.05.003.
13. Duchatelet S, Boyden LM, Ishida-Yamamoto A, Zhou J, Guibbal L, Hu R, et al. Mutations in PERP cause dominant and recessive keratoderma. J Invest Dermatol. 2019;139;380–390. https://doi.org/10.1016/J.JID.2018.08.026.
14. Wang H, Xu Z, Lee BH, Vu S, Hu L, Lee M, et al. Gain-of-function mutations in TRPM4 activation gate cause progressive symmetric erythrokeratodermia. J Invest Dermatol. 2019;139;1089–1097. https://doi.org/10.1016/J.JID.2018.10.044/ATTACHMENT/4A5430A4-C020-471F-B7C0-96992C1474D1/MMC1.PDF.
15. Van Steensel MAM, Oranje AP, van der Schroeff JG, Wagner A, van Geel M. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am J Med Genet A. 2009;149A;657–661. https://doi.org/10.1002/AJMG.A.32744.
16. Boyden LM, Craiglow BG, Hu RH, Zhou J, Browning J, Eichenfield L, et al. Phenotypic spectrum of autosomal recessive congenital ichthyosis due to PNPLA1 mutation. Br J Dermatol. 2017;177;319–322. https://doi.org/10.1111/BJD.15570.
17. Sun Q, Burgren NM, Cheraghlou S, Paller AS, Larralde M, Bercovitch L, et al. The genomic and phenotypic landscape of ichthyosis: an analysis of 1000 kindreds. JAMA Dermatol. 2022;158;16–25. https://doi.org/10.1001/JAMADERMATOL.2021.4242.
18. Dahlqvist J, Klar J, Tiwari N, Schuster J, Törmä H, Badhai J, et al. A single-nucleotide deletion in the POMP 5’ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am J Hum Genet. 2010;86;596–603. https://doi.org/10.1016/J.AJHG.2010.02.018.
According to the current classification of ichthyoses,8 there are 5 clinical forms of ARCI, including:
- -
Lamellar ichthyosis: characterized by a moderate-to-severe degree of erythema and desquamation in large lamellae of somewhat darker tone than the patient's skin, i.e. light brown in light phototypes and dark brown in darker phototypes. Although this large sheet-like desquamation affects the entire body, it is more evident on the lower extremities and the frontal region of the face (Fig. 5). Many patients also present ectropion and a thick adherent scalp desquamation that may precede an almost total cicatricial alopecia.26 Palmoplantar involvement also presents a laminar aspect, with areas of diffuse thickening and a smooth surface (Fig. 6). Alterations in nail morphology are not a rare finding, such as incurvation of the nail plate, linear areas of leukonychia and increased extension of the lunula.
- -
Congenital ichthyosiform erythroderma: together with lamellar ichthyosis, it is a classic phenotype of ARCI. Patients present with erythroderma of variable intensity and generalized desquamation with smaller scales than in the lamellar forms (Fig. 7). The scalp is usually less thick and adherent than in the lamellar forms. Characteristically, patients exhibit skin thickening of the dorsum of the hands and feet and a relative loss of elasticity of the skin of the fingers that determines a certain retraction of the fingers and a phenomenon of pseudoleukonychia when stretching them since cutaneous bed is exsanguinated (Fig. 8). Additionally, many patients also present a true leukonychia of probable post-inflammatory origin. Palmoplantar involvement determines a thickening of the palms and soles of leather-like appearance without true hyperkeratosis (Fig. 9). Many patients also exhibit, additionally, a striking facial reddening of the cheeks regardless of the severity of involvement of the rest of the body surface.
- -
Harlequin ichthyosis is the severe form of ARCI. It is so named because infants are born with rigid, thickened skin with extensive and deep linear fissures reminiscent of harlequin costumes. The rigidity of the skin integument is such that it restricts respiratory movements and sucking ability, endangering the child's life. Similarly, patients exhibit severe ectropion, eclabium, and severe limitation of the mobility of the fingers, which are enclosed by the corneous shell. This phenotype progresses weeks later to a severe form of congenital ichthyosiform erythroderma which, among other, associates symptoms, alopecia of the scalp, eyebrows and eyelashes, ectropion, anomalies of the auricular pavilions, joint deformities, and permanent mobility limitations27 (Fig. 10). It is due to recessive pathogenic variants that cause complete loss of function of ABCA12,27 with slightly less severe phenotypes in patients with 2 different heterozygous pathogenic variants.28
- -
Self-improving collodion baby: this is a clinical variant in which the collodion membrane progresses, regardless of its severity, to a mild ARCI phenotype. Although the expressivity of ichthyosis is very faint in some areas such as the trunk and proximal extremities, patients usually present with facial erythema, skin thickening on elbows, knees, and dorsum of hands and feet (Fig. 11), as well as palmoplantar thickening similar in appearance to that described in congenital ichthyosiform erythroderma. Phenotypic progression to self-improving forms is not predictable, which makes prognostic information of collodion infants within the first few days of life difficult to obtain. In fact, it is due to recessive pathogenic variants in ALOX12B,29ALOXE3,30CYP4F2231 or TGM1,32 genes that can also determine a much more important clinical involvement and therefore even an immediate molecular diagnosis cannot predict progression.
- -
Acral self-improving collodion baby is an exceptional clinical form in which the collodion membrane only affects the distal part of the extremities. It is due to recessive pathogenic variants in TGM1.33
- -
Bathing suit ichthyosis is an ARCI variant in which patients are born with a collodion membrane and progress to a lamellar ichthyosis phenotype of variable expressivity that only affects the trunk and scalp. It is an exceptional form initially described in the North African population34 and it is due to recessive pathogenic variants in TGM135 that generate a thermosensitive protein that loses activity in warmer areas of the body.







They receive the name keratinopathic ichthyoses because they are caused by pathogenic variants in genes encoding keratins, have a very low prevalence, estimated at 1.1 per million.24 Different clinical forms are distinguished according to the inheritance pattern and phenotypic features.
- -
Autosomal dominant keratinopathic ichthyosis: the most common form of keratinopathic ichthyosis. Its main characteristic is intercellular separation, which is why it is also called epidermolytic ichthyosis. Patients are born with erythroderma, blisters, and erosions so extensive that differential diagnosis with epidermolysis bullosa can be difficult to establish. As the weeks go by, patients suffer from less erosions and a diffuse hyperkeratosis grows, which will eventually become more accentuated on the flexor surfaces of the large folds and, later, on elbows and knees. Patients exhibit throughout their lives a variable degree of erythema and skin fragility that makes them prone to erosions with minimal trauma (Fig. 12). This group of ichthyoses is caused by dominant variants in KRT136 or KRT10.36 Those caused by variants in KRT1 commonly present palmoplantar keratoderma (Fig. 13), usually absent in those associated with pathogenic variants in KRT10, since in palmoplantar skin KRT9 can partially rescue the phenotype of a loss of KRT10.36
 Figure 12.
Figure 12.Epidermolytic keratinopathic ichthyosis due to a pathogenic variant in the KRT10 gene. Generalized involvement is observed with extensive areas of thickening in the upper and lower extremities and areas of skin fragility in the pretibial and lateral regions of the feet. The involvement stops at the palmoplantar border and spares the palms and soles.
(0.28MB). - -
Autosomal recessive keratinopathic ichthyosis: clinically very similar to the above. It is caused by pathogenic recessive variants in KRT10.37 Its prevalence is much lower than the dominant forms.
- -
Annular keratinopathic ichthyosis: a special subtype in which patients are born with an appearance similar to that of other keratinopathic ichthyoses, but subsequently progress to a less severe clinical form in which. In addition to diffuse hyperkeratosis at the level of the large folds, transient annular lesions with superficial desquamation are observed38 (Fig. 14). It is caused by dominant pathogenic variants in KRT139 or KRT10.40
- -
Superficial keratinopathic ichthyosis shows a milder phenotype than the 2 previous ones and does not present erythroderma at birth. It is characterized by mild-to-moderate diffuse hyperkeratosis showing predilection for articular surfaces. There may be focal detachment of the hyperkeratosis, a phenomenon called “mauserung” or “molting” in the international literature (Fig. 15). It is caused by dominant pathogenic variants in KRT2.41 This keratin is expressed in superficial layers of the stratum corneum and probably for this reason the clinical manifestation and the phenomena of epidermolysis and focal detachment of hyperkeratosis are less expressive vs cases related to pathogenic variants in KRT1 and KRT10.
- -
Mosaic keratinopathic ichthyoses are characterized by hyperkeratotic lesions of Blaschkoid distribution whose extent is variable, ranging from a few centimeters (epidermolytic nevus) to hemicorporal, uni or bilateral involvement. They are due to postzygotic pathogenic variants in KRT1,42KRT10,43 or KRT2,44 in which the disease-causing mutation only affects some epidermal progenitors. The area of affected skin varies depending on the timing of mutation, the earlier the mutation occurs, the more extensive it is. In cases where the mosaic affects the gonads, the disease can be transmitted to the patient's children as generalized keratinopathic ichthyosis.
- -
Keratinopathic ichthyosis with reversion patches (in confetti): a particular type of non-epidermolytic keratinopathic ichthyosis in which there is no epidermolysis and therefore the typical histological findings of intercellular separation are not observed. Clinically, patients present with severe ichthyosiform erythroderma and variable palmoplantar involvement. At school age, small lenticular areas of healthy skin begin to be observed on the trunk, which increase in number and size with age (Fig. 16). Patients may also present ectropion, scalp alopecia, nail changes, hypertrichosis, and abnormal gait, among other symptoms.45 It is caused by dominant variants in KRT1046 or, much less frequently, in KRT1, whose reversions appear in adulthood.47 Healthy skin patches are caused by loss of heterozygosity of the mutated allele due to mitotic recombination events whose triggering factor is unknown, but is thought to be related to aberrant import of mutated keratins to the nucleus.45
- -
Other non-syndromic ichthyoses are shown in Table 3. Although they are very rare, some have prominent clinical data that may facilitate diagnosis, such as superficial desquamation and underlying erythema in peeling skin syndromes (Fig. 17) or cribriform keratoderma produced by loricrin variants (Fig. 18).



 is observed.")






