





Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare cytotoxic T-cell lymphoma with indolent behavior, mostly present in women and associated with immunological diseases whose pathogenic background is still poorly understood. SPTCL is associated with lupus erythematosus panniculitis (LEP) and histologically misdiagnosed.
ObjectivesThe aim of our study was to identify mutations affecting the pathogenesis of both SPTCL and LEP.
Materials and methodsWe studied a total of 10 SPTCL and 10 LEP patients using targeted next-generation sequencing and pyrosequencing. Differences in gene expression between molecular subgroups were investigated using NanoString technology. Clinical data were collected, and correlations sought with the molecular data obtained.
ResultsThe mutational profile of SPTCL and LEP is different. We identified fewer pathogenic mutations than previously reported in SPTCL, noting a single HAVCR2-mutated SPTCL case. Interestingly, 40% of our SPTCL cases showed the pathogenic TP53 (p.Pro72Arg) (P72R) variant. Although cases showing HAVCR2 mutations or the TP53 (P72R) variant had more severe symptomatic disease, none developed hemophagocytic syndrome (HPS). Furthermore, TP53 (P72R)-positive cases were characterized by a lower metabolic signaling pathway and higher levels of CD28 expression and Treg signaling genes. In addition, 30% of our cases featured the same mutation (T735C) of the epigenetic modificatory gene DNMT3A. None of the LEP cases showed mutations in any of the studied genes.
ConclusionsThe mutational landscape of SPTCL is broader than previously anticipated. We describe, for the first time, the involvement of the TP53 (P72R) pathogenic variant in this subgroup of tumors, consider the possible role of different genetic backgrounds in the development of SPTCL, and conclude that LEP does not follow the same pathogenic pathway as SPTCL.
El linfoma T paniculítico (LTP) es un linfoma de células T citotóxico poco frecuente, de comportamiento indolente, más frecuente en mujeres, relacionado con enfermedades autoinmunes, y cuyos antecedentes patogénicos aún no se conocen bien. Se asocia y se confunde histológicamente con la paniculitis lúpica (PL).
ObjetivosEl objetivo de nuestro estudio fue identificar mutaciones implicadas en la patogénesis del LTP y de la PL.
Materiales y métodosSe estudiaron 10 pacientes con LTP y 10 con PL mediante secuenciación masiva (con un panel de genes customizados) y pirosecuenciación dirigida. Se investigaron diferencias en la expresión genética mediante NanoString entre diferentes subgrupos moleculares encontrados. Se recopilaron datos clínicos y se correlacionaron con los datos moleculares obtenidos.
ResultadosEl perfil mutacional del LTP y el de la LP son diferentes. El porcentaje de mutaciones encontradas en el subgrupo de LTP fue inferior al ya publicado en la literatura. Solo un paciente con LTP mostraba mutaciones en el gen HAVCR2. Curiosamente, el 40% de los LTP mostraron la variante patogénica TP53 (p.Pro72Arg) (P72R). Los pacientes con mutaciones en el gen HAVCR2 o con la variante TP53(P72R) sufrían enfermedad sintomática, aunque ninguno desarrolló síndrome hemofagocítico (SPH). El estudio de NanoString identificó que las muestras con alteración de TP53(P72R) se caracterizaban por una down-regulation de la vía de señalización del metabolismo y de una mayor expresión de los genes de las vías de señalización de CD28 y Treg si se comparaban con los casos negativos para TP53 (P72R). Además, el 30% de nuestros casos presentaban la misma mutación (T735C) en el gen modificador epigenético DNMT3A. Ninguno de los pacientes con PL mostró mutaciones en ninguno de los genes estudiados.
ConclusionesAmpliamos el perfil mutacional del LTP, describiendo por primera vez la implicación de la variante patogénica TP53 (P72R) en este subgrupo de tumores. Además, sugerimos el posible papel de un fondo genético en el desarrollo de los LTP. La aparición de PL no parece seguir la misma vía patogénica que la de los LTP.

Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare disease of TCR-αβ cytotoxic T-cells involving the subcutaneous tissue. In general, although SPTCL is an indolent disease, its association with the hemophagocytic syndrome (HPS) worsens prognosis.1,2 It is more common in women vs men and typically has an earlier age of onset than other T-cell lymphomas. SPTCL has a family background and is frequently associated with immunological diseases, especially lupus erythematosus.1,2 Differential diagnosis with LEP is sometimes challenging both clinically and histologically. Morphologically, the presence of follicles with germinal centers, plasma cells and clusters of plasmacytoid dendritic cells supports the diagnosis of LEP.2 Nevertheless, overlapping features of lupus erythematosus panniculitis (LEP) and SPTCL have been described in the same patient.3,4 On the other hand, gene-expression profiling of both diseases seemed to be different5 being overlapping histological cases closer to LEP profile vs SPTCL. These data suggest a different origin of both diseases.
Nevertheless, the pathogenic background of both LEP and SPTCL is still poorly understood. Regarding SPTCL, most series highlight the role of germline HAVCR2 gene mutations in its development.6,7 Other somatic changes in genes with epigenetic roles or involvement in the PI3K/AKT/mTOR or JAK/STAT signaling pathways with no recurrent hotspot mutations have been described too.8,9 Comparative genomic hybridization (CGH) studies have also shown a distinctive profile of losses and gains,10 and expression profiling studies have revealed differentially expressed genes and pathways.5,11 As far as we know, no known mutations associated with the development of LEP have ever been reported.
We conducted a comparative study on the mutational profile of 10 SPTCL cases and 10 LEP examples through targeted sequencing and pyrosequencing to determine whether these two entities shared a common pathogenic background.
Material and methodsPatients and samplesWe studied 10 SPTCL and 10 LEP biopsy specimens from patients diagnosed in various medical centers in Spain. Formalin-fixed, paraffin-embedded (FFPE) tissue sections from diagnostic biopsies were collected from 2006 through 2017. The research project was approved by Hospital Universitario Fundación Jimenez Díaz-IIS (CEIm-FJD) Ethics Committee (“Linfoma T paniculito y simuladores. Marcadores moleculares de diagnostico y terapia dirigida” PI17/02172. CI: 03/18) and conducted in full compliance with the Declaration of Helsinki. Further details are available in the supplementary data and methods. Histological features of these 20 cases were reviewed by two expert pathologists and dermatopathologists (SMRP and LR), respectively, and are already reported.5 No cases with overlapping features between SPTCL and LEP were included in this study.
ImmunohistochemistryThe immunohistochemical features of all these cases are already published. Moreover, apart from the conventional immunohistochemical markers needed to achieve diagnosis,5 P53 and FOXP3 were studied in all SPTCL cases. For the former, intense immunolabeling of, at least, 10% of the neoplastic cells for p53, or complete absence of p53 staining in neoplastic cells, was indicative of positivity. Expression of FOXP3 was quantified in the nucleus of bystander lymphoid cells and categorized into two groups: positive (≥50% positive cells) and negative (<50% positive cells).
Targeted next-generation sequencingGenomic DNA was extracted from FFPE tissue using a truXTRAC FFPE DNA Kit (Covaris, Woburn, MA, United States) following the manufacturer's instructions for use. A customized panel of 61 genes involved in lymphomagenesis-relevant pathways was designed using the SureDesign (Agilent Technologies, Santa Clara, CA, United States) web-based tool (supplementary Table 1 and supplementary data and methods).
PCR primer design and PCR amplificationPCR-specific primers were designed using the Entrez Global Query Cross-Database Search System and the Basic Local Alignment Search Tool (BLAST) of the National Center for Biotechnology Information (NCBI) website and the Lasergene sequence analysis software (DNAStar, Lasergene®). Specific amplification primers were designed for amplifying HAVCR2 (the gene encoding TIM3) [GenBank: NG_030444.1, NM_032782.3, NP_116171.3] (supplementary Figure 1 and supplementary data and methods).
nCounter gene-expression assayAfter histological practical revision and quality control, gene-expression assays of 10 cases were successfully conducted. Gene-expression profiling (GEP) was performed with nCounter Technology (NanoString Technologies, Seattle, WA, United States). Total RNA from FFPE sections was isolated from diagnostic samples using a truXTRAC FFPE Total NA Kit (Covaris Inc., Woburn, MA, United States) following the manufacturer's instructions for use.5
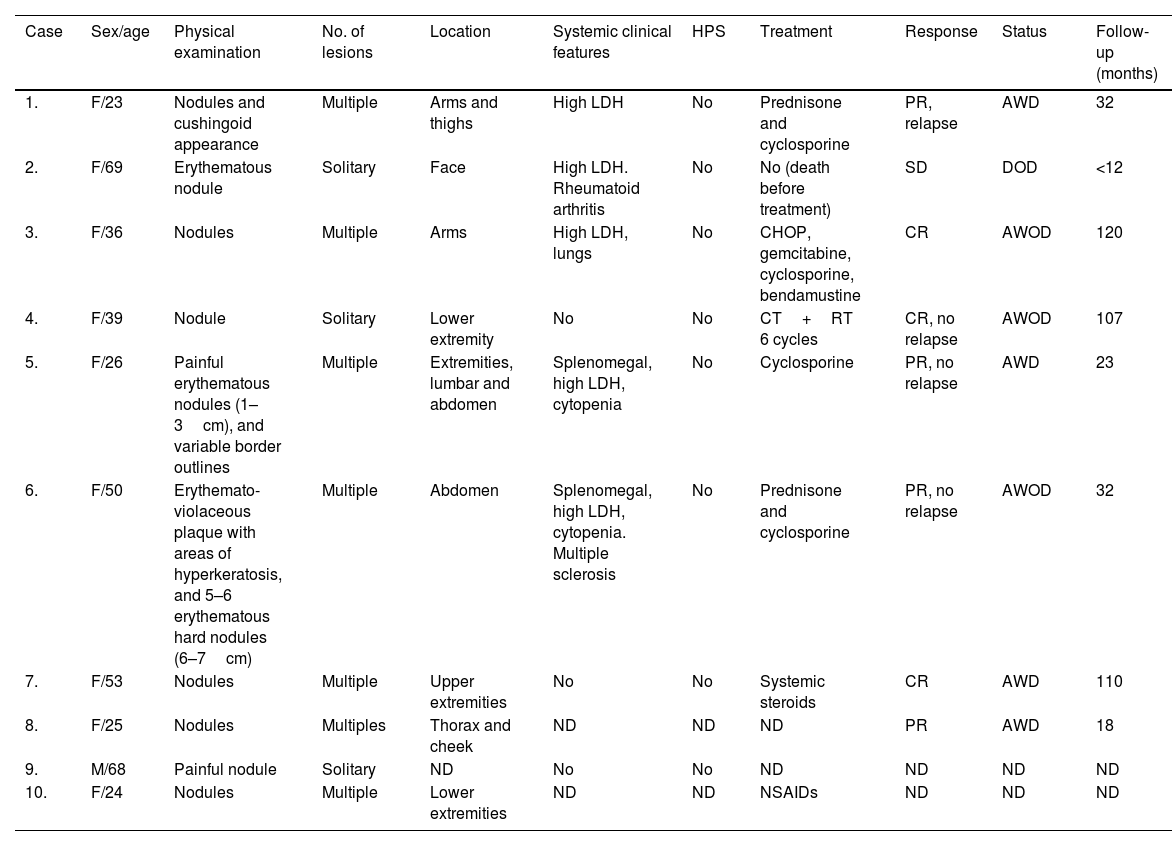
ResultsThe clinical findings of these SPTCL series are summarized in Table 1. Nive out of the 10 patients with SPTCL were female vs 1 man. The mean and median ages at diagnosis were 41.3 and 37.5 years, respectively. Solitary (3/10) or multiple (7/10) plaques or nodules of SPTCL were located on the upper extremities (4/9), lower extremities (4/9), face (2/9) and/or trunk (3/9) (1 patient's location was not in the records). Four patients were on immunomodulators, two on chemotherapy, one on non-steroidal anti-inflammatory drugs, and one patient died before receiving any therapies (case #2). High levels of LDH and cytopenia were observed in five and two patients, respectively. None of our cases presented HPS. Excluding the patient who died of an unrelated disease before treatment, all patients were alive with the active disease (four patients) or disease-free (three patients) at the last follow-up, which ranged from 1 to 10 years.
Clinical features of subcutaneous panniculitis-like T-cell lymphoma cases.
| Case | Sex/age | Physical examination | No. of lesions | Location | Systemic clinical features | HPS | Treatment | Response | Status | Follow-up (months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | F/23 | Nodules and cushingoid appearance | Multiple | Arms and thighs | High LDH | No | Prednisone and cyclosporine | PR, relapse | AWD | 32 |
| 2. | F/69 | Erythematous nodule | Solitary | Face | High LDH. Rheumatoid arthritis | No | No (death before treatment) | SD | DOD | <12 |
| 3. | F/36 | Nodules | Multiple | Arms | High LDH, lungs | No | CHOP, gemcitabine, cyclosporine, bendamustine | CR | AWOD | 120 |
| 4. | F/39 | Nodule | Solitary | Lower extremity | No | No | CT+RT 6 cycles | CR, no relapse | AWOD | 107 |
| 5. | F/26 | Painful erythematous nodules (1–3cm), and variable border outlines | Multiple | Extremities, lumbar and abdomen | Splenomegal, high LDH, cytopenia | No | Cyclosporine | PR, no relapse | AWD | 23 |
| 6. | F/50 | Erythemato-violaceous plaque with areas of hyperkeratosis, and 5–6 erythematous hard nodules (6–7cm) | Multiple | Abdomen | Splenomegal, high LDH, cytopenia. Multiple sclerosis | No | Prednisone and cyclosporine | PR, no relapse | AWOD | 32 |
| 7. | F/53 | Nodules | Multiple | Upper extremities | No | No | Systemic steroids | CR | AWD | 110 |
| 8. | F/25 | Nodules | Multiples | Thorax and cheek | ND | ND | ND | PR | AWD | 18 |
| 9. | M/68 | Painful nodule | Solitary | ND | No | No | ND | ND | ND | ND |
| 10. | F/24 | Nodules | Multiple | Lower extremities | ND | ND | NSAIDs | ND | ND | ND |
AWD, alive with disease; AWOD, alive without disease; CT, chemotherapy; CR, complete response; DOD, dead of other disease; ND, not determined; NSAIDs, non-steroidal anti-inflammatory drugs; PR, partial response; RT, radiotherapy; SD, stable disease.
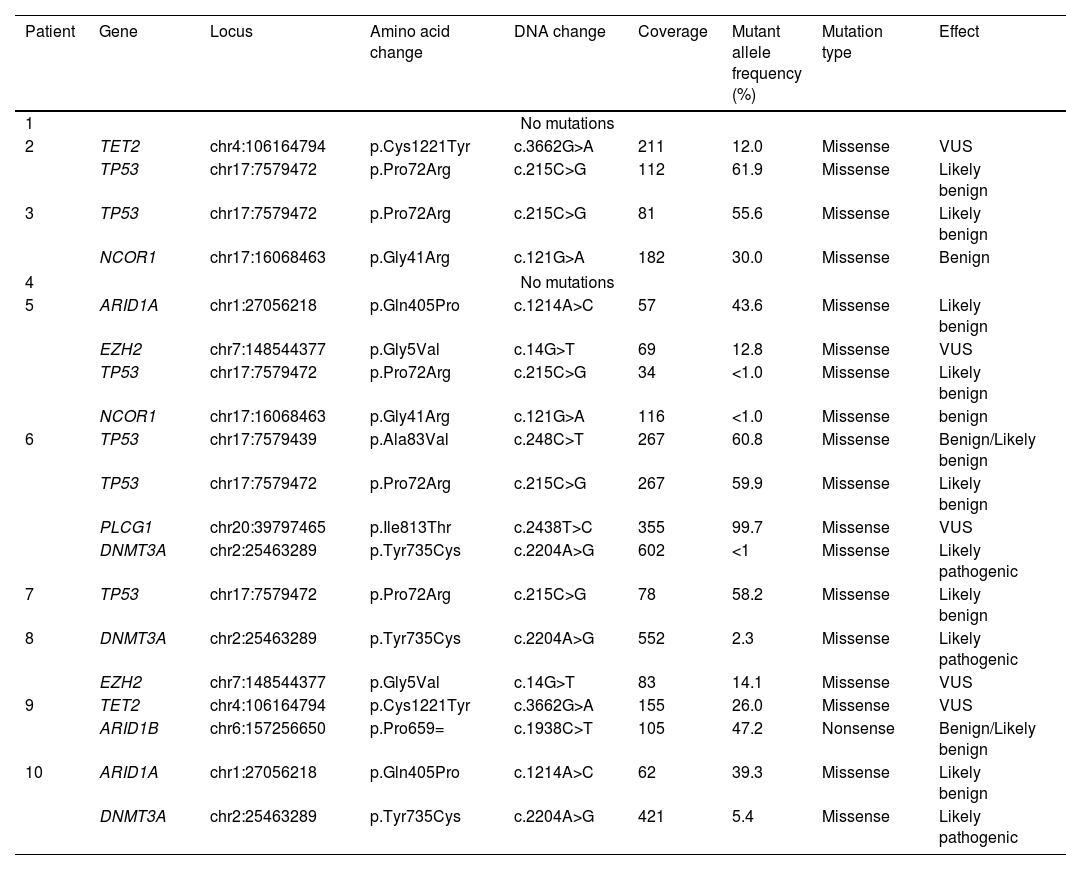
Using next-generation sequencing (NGS) we found that TP53 was altered in the proline-rich domain (PRD) (Figs. 1 and 2) in 50% (5/10) of the cases, making this the most frequent alteration in the patients studied (Table 2). Residue 72 was recurrently mutated (P72R) in these five cases. No correlation was ever found between the presence/absence of this variant and the level of protein expression. Conversely, differences were found in the expression profile between TP53 (P72R) variant-positive and variant-negative cases identified using the NanoString technique (supplementary Figures 2 and 3). The TP53 (P72R) variant-positive cases overexpressed CD28 and Treg signaling pathways and their metabolic pathway was downregulated vs the TP53 (P72R) variant-negative cases. The level of FOXP3 expression was higher in the TP53 (P72R) variant-positive subgroup (supplementary Table 2). Three out of the 5 TP53 (P72R)-positive cases showed high levels of LDH and systemic symptoms.


Mutations detected by next-generation sequencing in our cases of subcutaneous panniculitis-like T-cell lymphoma.
| Patient | Gene | Locus | Amino acid change | DNA change | Coverage | Mutant allele frequency (%) | Mutation type | Effect |
|---|---|---|---|---|---|---|---|---|
| 1 | No mutations | |||||||
| 2 | TET2 | chr4:106164794 | p.Cys1221Tyr | c.3662G>A | 211 | 12.0 | Missense | VUS |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 112 | 61.9 | Missense | Likely benign | |
| 3 | TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 81 | 55.6 | Missense | Likely benign |
| NCOR1 | chr17:16068463 | p.Gly41Arg | c.121G>A | 182 | 30.0 | Missense | Benign | |
| 4 | No mutations | |||||||
| 5 | ARID1A | chr1:27056218 | p.Gln405Pro | c.1214A>C | 57 | 43.6 | Missense | Likely benign |
| EZH2 | chr7:148544377 | p.Gly5Val | c.14G>T | 69 | 12.8 | Missense | VUS | |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 34 | <1.0 | Missense | Likely benign | |
| NCOR1 | chr17:16068463 | p.Gly41Arg | c.121G>A | 116 | <1.0 | Missense | benign | |
| 6 | TP53 | chr17:7579439 | p.Ala83Val | c.248C>T | 267 | 60.8 | Missense | Benign/Likely benign |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 267 | 59.9 | Missense | Likely benign | |
| PLCG1 | chr20:39797465 | p.Ile813Thr | c.2438T>C | 355 | 99.7 | Missense | VUS | |
| DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 602 | <1 | Missense | Likely pathogenic | |
| 7 | TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 78 | 58.2 | Missense | Likely benign |
| 8 | DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 552 | 2.3 | Missense | Likely pathogenic |
| EZH2 | chr7:148544377 | p.Gly5Val | c.14G>T | 83 | 14.1 | Missense | VUS | |
| 9 | TET2 | chr4:106164794 | p.Cys1221Tyr | c.3662G>A | 155 | 26.0 | Missense | VUS |
| ARID1B | chr6:157256650 | p.Pro659= | c.1938C>T | 105 | 47.2 | Nonsense | Benign/Likely benign | |
| 10 | ARID1A | chr1:27056218 | p.Gln405Pro | c.1214A>C | 62 | 39.3 | Missense | Likely benign |
| DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 421 | 5.4 | Missense | Likely pathogenic | |
VUS, variant of unknown significance.
Mutations that were probably pathogenic in epigenetic-related genes (TET2, DNMT3A, EZH2, ARID1A, ARID1B, NCOR) were found in only 30% of SPTCL patients (cases #6, #8, and #10). These three patients had the same variant Y735C in DNMT3A. The change was in the methyltransferase domain of the gene (Fig. 2).
Pyrosequencing revealed that only one case (case 35) – a 26-year-old woman with systemic symptoms without HPS) – had the HAVCR2 (Y82C) mutation in homozygosis (Fig. 3).

None of the LEP cases showed any of these mutations, including the TP53 (P72R) variant.
DiscussionData presented here suggest the presence ethiopathogenic differences between SPTCL and LEP. Further high-throughput technologies should be used for LEP background understanding. Regarding SPTCL, our data differs from former studies.
Germline mutation of the hepatitis A virus-cellular receptor 2 (HAVCR2, which encodes T-cell immunoglobulin and mucin domain-containing protein 3 [TIM-3]) seems to be the hallmark of SPTCL.7 It has been previously reported in a high number of patients with sporadic SPTCL, ranging from 25% up to 85% of the cases analyzed.6,7,12,13 The lowest percentage of positive cases was reported in the European series by Sonigo et al.12 Thirteen of the 53 cases could be analyzed, only three of whom were of European origin (i.e., 23% of the positive cases and 6% of the whole series). Only one patient (10%, 1/10) from our series, and 0 from the 10 LEP cases studied featured mutated SPTCL. These data suggest a different background for the Asian and European patients with respect to the development of SPTCLs. The impact of genetic background, and that of microenvironmental factors, on the worldwide distribution of other subgroups of SPTCL has been reported previously.14–16
Three major mutations have been reported in the HAVCR2 gene as closely associated with the patients’ ethnic background: the Y82C and T101I variants, which are associated with Asian and Polynesian ancestry, and I97M, which has been found most frequently in patients with a North African or Caucasian background. We should also mention that the I97M variant has been reported only very rarely in Asian populations,7 while the Y82C variant has been found only occasionally in populations different than the Asian one, in most instances in heterozygosity.6,12 In fact, only one case of the Y82C variant mutation of the HAVCR2 gene in homozygosis has been found in one South American patient.6 Interestingly, we found the Y82C variant in homozygosis in a 26-year-old Spanish woman with no known foreign family background or previous family history of either lymphoma or autoimmune disease. Patients with HAVCR2 mutations have a younger median age at disease onset, male sex predominance, a longer median time to diagnosis, a more severe course of the disease, a higher rate of autoantibodies and more chances of systemic illness and HPS development.6,13,17,18 Moreover, HAVCR2-mutated tumors are rich in inflammatory, IL6-JAK-STAT3, TNF-α and NFK-B signaling pathways. Conversely, non-mutated cases are highlighted by a lymphocyte homing and an autoimmune profile. Our HAVCR2-mutated patient showed systemic symptoms but not HPS. In fact, none of the patients studied in this series exhibited HPS.
Differences from former studies may be associated with differences of ethnicity, age of disease onset, the family background of lymphoma disease and the percentage of cases with HPS in the present series.
We reported, for the first time ever, the presence of the TP53 (P72R) gene mutation in SPTCLs. It has been reported that this mutation has is a common single-nucleotide polymorphism (SNP) with a significant ethnic bias, whereby it is present in homozygosis in up to 40% of Caucasian Americans vs only ∼8% of African Americans. However, it was not present in any of the 10 LEPs analyzed. Although this SNP is not supposed to influence cancer risk, it has been associated with increased weight, type 2 diabetes risk, and inflammation.19 This inflammatory property has been involved in the increase of several subtypes of tumor aggressiveness.20
Phenotypic differences were found between the TP53 (P72R) variant-positive and -negative SPTCL cases. The TP53 (P72R) variant-positive cases featured upregulation and downregulation of the Treg/CD28 and metabolic pathways, respectively. A close relationship among CD28, TNF, and Treg development has been reported.21 An increase in TNF and a lower metabolic activity have been reported in R72 transgenic mice.19 Moreover, p53 has proven to be an important modulator of CD4+ T-cell differentiation including Th17 cells and Tregs.22 No differences in p53 protein expression were found between the TP53 (P72R) variant-positive and -negative cases. Furthermore, Tregs have been proposed as the “key players” in the crosstalk between metabolisms and immunity. An inverse correlation between leptin levels (an adipocytokine produced by adipose tissue in response to the amount of fat) and the abundance of Treg cell in autoimmune diseases has been reported.23 High levels of leptin have been reported in systemic lupus erythematosus (SLE),24 and a meta-analysis has reported that the p53 activity does not seem to correlate with the pathogenesis of that disease.24
Interestingly, the mutations detected here could be useful targets regarding treatment. Several strategies targeting oncogenic mutant TP53 in cancers exist. Polymorphism in codon 72 (Arg/Pro) of TP53, the transcription factor encoded by TP53, affects cellular sensitivity to anticancer drugs such as doxorubicin through inhibition of p73, a protein related to p53.25 Furthermore, drugs targeting epigenetic changes have shown promising results.26–28 Romidepsin (a HDAC inhibitor)8 has recently demonstrated to be an effective therapy for subcutaneous panniculitis-like T-cell lymphoma, giving a complete response as monotherapy in treatment-resistant cases of the disease.29
Our study expands the mutational landscape of SPTCL, suggesting that there is an important ethnic background underlying the development of this disease, and highlighting the relevance of different molecular backgrounds in the development of SPTCL and LEP. However, our series is limited and further studies in a larger case series is needed to validate our findings.
Conflicts of interestNone declared.
This work was supported by grants from the Instituto de Salud Carlos III, from the Ministry of Science and Innovation of Spain, PI17/2172; ISCIII-MINECO-AES-FEDER. R.A.-A. is the recipient of PFIS predoctoral fellowship. L.T.-R. is funded by Marie Skłodowska-Curie Individual Fellowship (No. 882597). M.R.-M. is supported by CIBERONC (CB16/12/00291). P.M. has a Miguel Servet contract funded by the ISCIII (CP16/00116). L.d.l.F. was supported by the ISCIII contract CA18/00017.
The following are the supplementary data to this article:



