




El linfoma T paniculítico (LTP) es un linfoma de células T citotóxico poco frecuente, de comportamiento indolente, más frecuente en las mujeres, relacionado con enfermedades autoinmunes, y cuyos antecedentes patogénicos aún no se conocen bien. Se asocia y se confunde histológicamente con la paniculitis lúpica (PL).
ObjetivosEl objetivo de nuestro estudio fue identificar mutaciones implicadas en la patogénesis del LTP y de la PL.
Materiales y métodosSe estudiaron 10 pacientes con LTP y 10 con PL mediante secuenciación masiva (con un panel de genes customizados) y pirosecuenciación dirigida. Se investigaron diferencias en la expresión genética mediante NanoString® entre diferentes subgrupos moleculares encontrados. Se recopilaron los datos clínicos, y se correlacionaron con los datos moleculares obtenidos.
ResultadosEl perfil mutacional del LTP y el de la LP son diferentes. El porcentaje de mutaciones encontradas en el subgrupo de LTP fue inferior al ya publicado en la literatura. Solo un paciente con LTP mostraba mutaciones en el gen HAVCR2. Curiosamente, el 40% de los LTP mostraron la variante patogénica TP53 (p.Pro72Arg) (P72R). Los pacientes con mutaciones en el gen HAVCR2 o con la variante TP53(P72R) sufrían enfermedad sintomática, aunque ninguno desarrolló síndrome hemofagocítico (SPH). El estudio de NanoString® identificó que las muestras con alteración de TP53(P72R) se caracterizaban por una down-regulation de la vía de señalización del metabolismo y de una mayor expresión de los genes de las vías de señalización de CD28 y Treg si se comparaban con los casos negativos para TP53 (P72R). Además, el 30% de nuestros casos presentaban la misma mutación (T735C) en el gen modificador epigenético DNMT3A. Ninguno de los pacientes con PL mostró mutaciones en ninguno de los genes estudiados.
ConclusionesAmpliamos el perfil mutacional del LTP, describiendo por primera vez la implicación de la variante patogénica TP53(P72R) en este subgrupo de tumores. Además, sugerimos el posible papel de un fondo genético en el desarrollo de los LTP. La aparición de PL no parece seguir la misma vía patogénica que la de los LTP.
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare cytotoxic T-cell lymphoma with indolent behavior, mostly present in women and associated with immunological diseases whose pathogenic background is still poorly understood. SPTCL is associated with lupus erythematosus panniculitis (LEP) and histologically misdiagnosed.
ObjectivesThe aim of our study was to identify mutations affecting the pathogenesis of both SPTCL and LEP.
Materials and methodsWe studied a total of 10 SPTCL and 10 LEP patients using targeted next-generation sequencing and pyrosequencing. Differences in gene expression between molecular subgroups were investigated using NanoString® technology. Clinical data were collected, and correlations sought with the molecular data obtained.
ResultsThe mutational profile of SPTCL and LEP is different. We identified fewer pathogenic mutations than previously reported in SPTCL, noting a single HAVCR2-mutated SPTCL case. Interestingly, 40% of our SPTCL cases showed the pathogenic TP53 (p.Pro72Arg) (P72R) variant. Although cases showing HAVCR2 mutations or the TP53(P72R) variant had more severe symptomatic disease, none developed hemophagocytic syndrome (HPS). Furthermore, TP53(P72R)-positive cases were characterized by a lower metabolic signaling pathway and higher levels of CD28 expression and Treg signaling genes. In addition, 30% of our cases featured the same mutation (T735C) of the epigenetic modificatory gene DNMT3A. None of the LEP cases showed mutations in any of the studied genes.
ConclusionsThe mutational landscape of SPTCL is broader than previously anticipated. We describe, for the first time, the involvement of the TP53(P72R) pathogenic variant in this subgroup of tumors, consider the possible role of different genetic backgrounds in the development of SPTCL, and conclude that LEP does not follow the same pathogenic pathway as SPTCL.

El linfoma subcutáneo de células T de tipo paniculitis (SPTCL) es una enfermedad rara de células T citotóxicas TCR-ab que afecta al tejido subcutáneo. En general, aunque el SPTCL es una enfermedad indolente, su asociación con el síndrome hemofagocítico (HPS) empeora su pronóstico1,2. Es más frecuente en las mujeres que en los varones, y suele tener una edad de aparición más temprana que otros linfomas de células T. En el SPTCL existen antecedentes familiares y, además, este se asocia con frecuencia a enfermedades inmunológicas, especialmente el lupus eritematoso1,2. El diagnóstico diferencial con la paniculitis lúpica eritematosa (LEP) es a veces difícil tanto clínica como histológicamente. Morfológicamente, la presencia de folículos con centros germinales, células plasmáticas y grupos de células dendríticas plasmocitoides apoya el diagnóstico de LEP2. Sin embargo, se han descrito características superpuestas de LEP y SPTCL en el mismo paciente3,4. Por otro lado, el perfil de expresión génica de ambas enfermedades parece ser diferente5, siendo que los casos que presentan características histológicas superpuestas se aproximaran más al perfil del LEP que al del SPTCL. Estos datos sugieren un origen diferente en ambas enfermedades.
No obstante, el trasfondo patogénico tanto del LEP como del SPTCL sigue siendo poco conocido. En cuanto al SPTCL, la mayoría de las series destacan el papel de las mutaciones del gen HAVCR2 en la línea germinal de su patogenia6,7. También se han descrito otros cambios somáticos en genes con funciones epigenéticas o implicados en las vías de señalización PI3K/AKT/mTOR o JAK/STAT sin mutaciones recurrentes en puntos críticos8,9.
Los estudios de hibridación genómica comparada (CGH) también han mostrado un perfil distinto de pérdidas y ganancias10. Así mismo, los estudios de los perfiles de expresión han revelado la existencia de genes y de vías que se expresan de forma distinta en ambas entidades5,11. Hasta donde sabemos, hasta el momento no se han descrito mutaciones asociadas al desarrollo del LEP.
Realizamos un estudio comparativo del perfil mutacional de 10 casos de SPTCL y 10 ejemplos de LEP mediante secuenciación dirigida y pirosecuenciación para determinar si estas 2 entidades compartían un trasfondo patogénico común.
Material y métodosPacientes y muestrasSe estudiaron 10 muestras de biopsias de SPTCL y 10 de LEP de pacientes diagnosticados en diversos centros de España. Se recogieron secciones de tejido fijadas en formol e incluidas en parafina (FFPE) procedentes de biopsias diagnósticas realizadas entre el año 2006 hasta el 2017. El proyecto de investigación fue aprobado por el Comité de Ética del Hospital Universitario Fundación Jiménez Díaz-IIS (CEIm-FJD) («Linfoma T paniculítico y simuladores. Marcadores moleculares de diagnóstico y terapia dirigida» PI17/02172. CI: 03/18) y realizado en total conformidad con la Declaración de Helsinki. Para más detalles, consúltese el Apéndice Datos y métodos suplementarios. Las características histológicas de estos 20 casos fueron revisadas por 2 patólogos y dermatopatólogos expertos (SMRP y LR), respectivamente, y ya fueron previamente presentados5. En el presente estudio no se han incluido casos con características que coincidieran entre el SPTCL y el LEP.
InmunohistoquímicaLas características inmunohistoquímicas de todos estos casos ya han sido publicadas. Además, aparte de los marcadores inmunohistoquímicos convencionales necesarios para alcanzar el diagnóstico5, se estudiaron el P53 y el FOXP3 en todos los casos de SPTCL. Para el primero, la inmunomarcación intensa de, al menos, el 10% de las células neoplásicas para el p53, o la ausencia completa de tinción para el p53 en las células neoplásicas, era indicativa de positividad. La expresión del FOXP3 se cuantificó en el núcleo de las células linfoides y se clasificó en 2 grupos: positivos (≥50% de células positivas) y negativos (<50% de células positivas).
Secuenciación de nueva generación dirigidaEl ADN genómico se extrajo de tejido FFPE utilizando un kit de ADN FFPE truXTRAC® (Covaris, Woburn, MA, EE. UU.) siguiendo las instrucciones de uso del fabricante. Se diseñó un panel personalizado de 61 genes implicados en las vías relevantes para la linfomagénesis utilizando la herramienta basada en la web SureDesign (Agilent Technologies, Santa Clara, CA, EE. UU.) (Apéndice tabla suplementaria 1 y datos y métodos suplementarios).
Diseño de cebadores PCR y amplificación PCRLos cebadores específicos para la PCR se diseñaron utilizando el Entrez Global Query Cross-Database Search System y la Basic Local Alignment Search Tool (BLAST) del sitio web del National Center for Biotechnology Information (NCBI) y el software de análisis de secuencias Lasergene (DNAStar, Lasergene®). Se diseñaron cebadores de amplificación específicos para amplificar HAVCR2 (el gen que codifica TIM3) [GenBank: NG_030444.1, NM_032782.3, NP_116171.3] (Apéndice figura suplementaria 1 y datos y métodos suplementarios).
Ensayo de expresión génica nCounterTras la revisión práctica histológica y el control de calidad, se realizaron con éxito los ensayos de expresión génica de 10 casos. El perfil de expresión génica (GEP) se realizó con la tecnología nCounter (NanoString® Technologies, Seattle, WA, EE. UU.). El ARN total de secciones FFPE se aisló a partir de muestras de diagnóstico utilizando un kit truXTRAC FFPE Total NA (Covaris Inc., Woburn, MA, EE. UU.) siguiendo las instrucciones de uso del fabricante5.
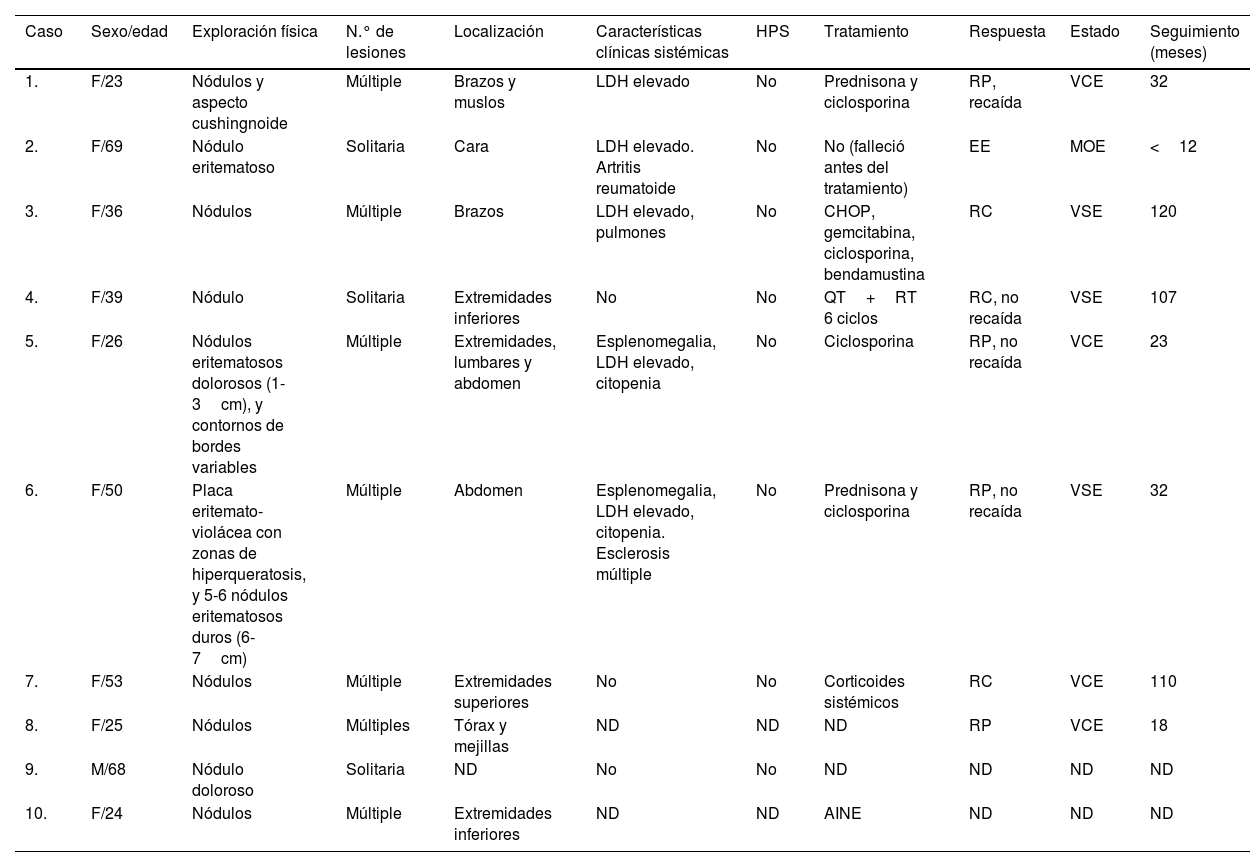
ResultadosLos hallazgos clínicos de estas series de SPTCL se resumen en la tabla 1. Nueve de los 10 pacientes con SPTCL eran mujeres frente a un varón. Las edades media y mediana al diagnóstico fueron de 41,3 y 37,5 años, respectivamente. Las placas o nódulos solitarios (3/10) o múltiples (7/10) de SPTCL se localizaron en las extremidades superiores (4/9), extremidades inferiores (4/9), cara (2/9) y/o tronco (3/9) (la localización de un paciente no constaba en los registros). Cuatro pacientes recibieron inmunomoduladores, 2 recibieron quimioterapia, uno antiinflamatorios no esteroideos y un paciente falleció antes de haber recibido algún tratamiento (caso n.° 2). Se observaron niveles elevados de LDH y citopenia en 5 y 2 pacientes, respectivamente. Ninguno de nuestros casos presentó HPS. Excluyendo al paciente que murió antes de haber recibido el tratamiento debido a una enfermedad no relacionada, todos los pacientes estaban vivos con la enfermedad activa (4 pacientes) o libres de enfermedad (3 pacientes) en el último seguimiento, que osciló entre uno y 10 años.
Características clínicas de los casos de linfoma subcutáneo de células T de tipo paniculitis
| Caso | Sexo/edad | Exploración física | N.° de lesiones | Localización | Características clínicas sistémicas | HPS | Tratamiento | Respuesta | Estado | Seguimiento (meses) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | F/23 | Nódulos y aspecto cushingnoide | Múltiple | Brazos y muslos | LDH elevado | No | Prednisona y ciclosporina | RP, recaída | VCE | 32 |
| 2. | F/69 | Nódulo eritematoso | Solitaria | Cara | LDH elevado. Artritis reumatoide | No | No (falleció antes del tratamiento) | EE | MOE | <12 |
| 3. | F/36 | Nódulos | Múltiple | Brazos | LDH elevado, pulmones | No | CHOP, gemcitabina, ciclosporina, bendamustina | RC | VSE | 120 |
| 4. | F/39 | Nódulo | Solitaria | Extremidades inferiores | No | No | QT+RT 6 ciclos | RC, no recaída | VSE | 107 |
| 5. | F/26 | Nódulos eritematosos dolorosos (1-3cm), y contornos de bordes variables | Múltiple | Extremidades, lumbares y abdomen | Esplenomegalia, LDH elevado, citopenia | No | Ciclosporina | RP, no recaída | VCE | 23 |
| 6. | F/50 | Placa eritemato-violácea con zonas de hiperqueratosis, y 5-6 nódulos eritematosos duros (6-7cm) | Múltiple | Abdomen | Esplenomegalia, LDH elevado, citopenia. Esclerosis múltiple | No | Prednisona y ciclosporina | RP, no recaída | VSE | 32 |
| 7. | F/53 | Nódulos | Múltiple | Extremidades superiores | No | No | Corticoides sistémicos | RC | VCE | 110 |
| 8. | F/25 | Nódulos | Múltiples | Tórax y mejillas | ND | ND | ND | RP | VCE | 18 |
| 9. | M/68 | Nódulo doloroso | Solitaria | ND | No | No | ND | ND | ND | ND |
| 10. | F/24 | Nódulos | Múltiple | Extremidades inferiores | ND | ND | AINE | ND | ND | ND |
AINE: antiinflamatorios no esteroideos; EE: enfermedad estable; MOE: muerto por otra enfermedad; ND: no determinado; QT: quimioterapia; RC: respuesta completa; RP: respuesta parcial; RT: radioterapia; VCE: vivo con enfermedad; VSE: vivo sin enfermedad.
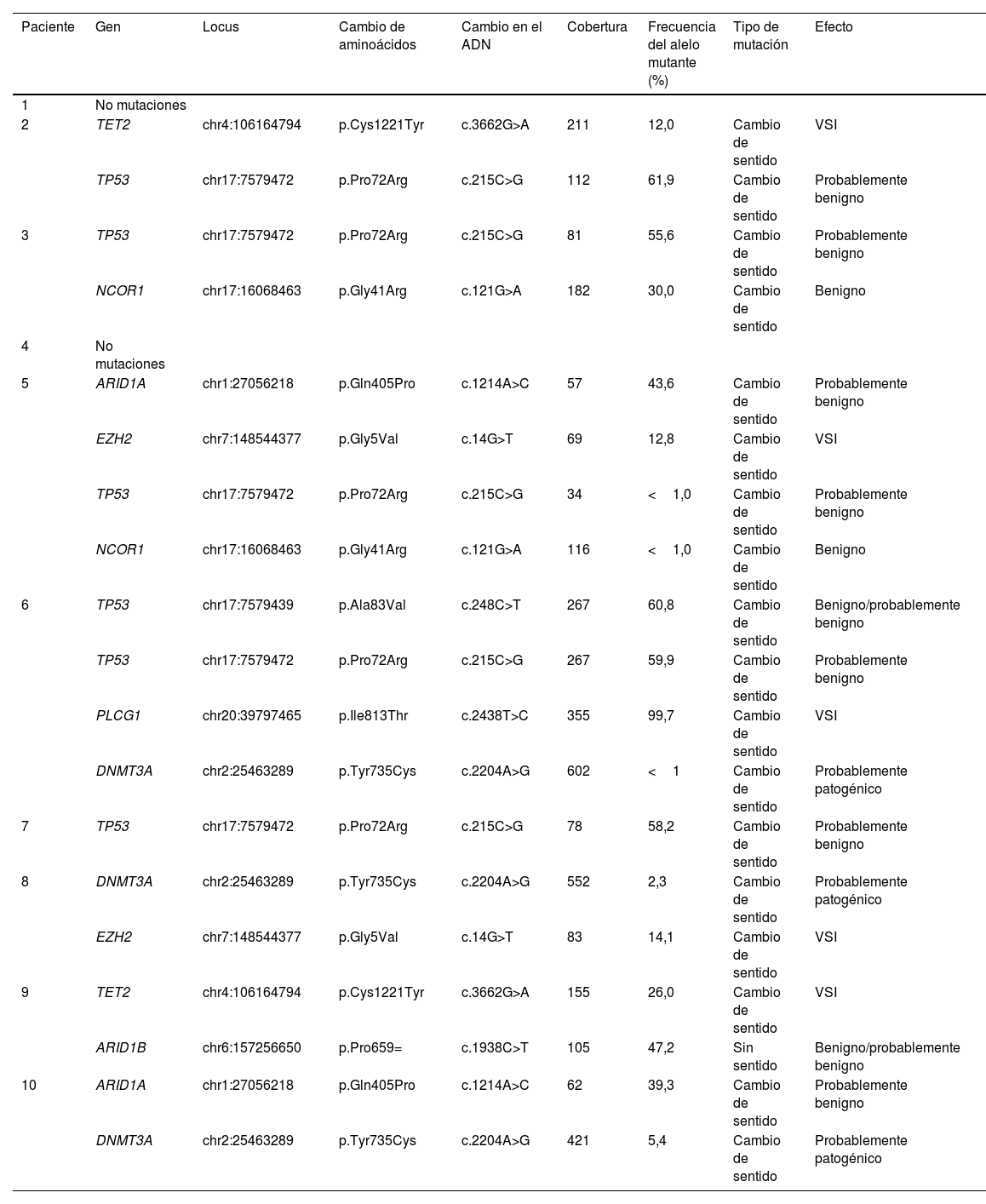
Mediante secuenciación de nueva generación (NGS) encontramos que el TP53 estaba alterado en el dominio rico en prolina (PRD) (figs. 1 y 2) en el 50% (5/10) de los casos, siendo esta la alteración más frecuente en los pacientes estudiados (tabla 2).


Mutaciones detectadas mediante secuenciación de nueva generación en nuestros casos de linfoma subcutáneo de células T similares a paniculitis
| Paciente | Gen | Locus | Cambio de aminoácidos | Cambio en el ADN | Cobertura | Frecuencia del alelo mutante (%) | Tipo de mutación | Efecto |
|---|---|---|---|---|---|---|---|---|
| 1 | No mutaciones | |||||||
| 2 | TET2 | chr4:106164794 | p.Cys1221Tyr | c.3662G>A | 211 | 12,0 | Cambio de sentido | VSI |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 112 | 61,9 | Cambio de sentido | Probablemente benigno | |
| 3 | TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 81 | 55,6 | Cambio de sentido | Probablemente benigno |
| NCOR1 | chr17:16068463 | p.Gly41Arg | c.121G>A | 182 | 30,0 | Cambio de sentido | Benigno | |
| 4 | No mutaciones | |||||||
| 5 | ARID1A | chr1:27056218 | p.Gln405Pro | c.1214A>C | 57 | 43,6 | Cambio de sentido | Probablemente benigno |
| EZH2 | chr7:148544377 | p.Gly5Val | c.14G>T | 69 | 12,8 | Cambio de sentido | VSI | |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 34 | <1,0 | Cambio de sentido | Probablemente benigno | |
| NCOR1 | chr17:16068463 | p.Gly41Arg | c.121G>A | 116 | <1,0 | Cambio de sentido | Benigno | |
| 6 | TP53 | chr17:7579439 | p.Ala83Val | c.248C>T | 267 | 60,8 | Cambio de sentido | Benigno/probablemente benigno |
| TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 267 | 59,9 | Cambio de sentido | Probablemente benigno | |
| PLCG1 | chr20:39797465 | p.Ile813Thr | c.2438T>C | 355 | 99,7 | Cambio de sentido | VSI | |
| DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 602 | <1 | Cambio de sentido | Probablemente patogénico | |
| 7 | TP53 | chr17:7579472 | p.Pro72Arg | c.215C>G | 78 | 58,2 | Cambio de sentido | Probablemente benigno |
| 8 | DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 552 | 2,3 | Cambio de sentido | Probablemente patogénico |
| EZH2 | chr7:148544377 | p.Gly5Val | c.14G>T | 83 | 14,1 | Cambio de sentido | VSI | |
| 9 | TET2 | chr4:106164794 | p.Cys1221Tyr | c.3662G>A | 155 | 26,0 | Cambio de sentido | VSI |
| ARID1B | chr6:157256650 | p.Pro659= | c.1938C>T | 105 | 47,2 | Sin sentido | Benigno/probablemente benigno | |
| 10 | ARID1A | chr1:27056218 | p.Gln405Pro | c.1214A>C | 62 | 39,3 | Cambio de sentido | Probablemente benigno |
| DNMT3A | chr2:25463289 | p.Tyr735Cys | c.2204A>G | 421 | 5,4 | Cambio de sentido | Probablemente patogénico | |
VSI: variante de significado incierto.
El residuo 72 estaba mutado de forma recurrente (P72R) en estos 4 casos. No se encontró correlación entre la presencia/ausencia de esta variante y el nivel de expresión proteica. Por el contrario, se encontraron diferencias en el perfil de expresión entre los casos con variante de TP53(P72R) positiva y los casos con variante negativa identificados mediante la técnica NanoString® (Apéndice figuras suplementarias 2 y 3). Los casos positivos para la variante TP53(P72R) sobreexpresaban las vías de señalización CD28 y Treg y su vía metabólica estaba regulada a la baja frente a los casos negativos para la variante de TP53 (P72R). El nivel de expresión de FOXP3 fue mayor en el subgrupo positivo para la variante TP53(P72R) (Apéndice, tabla suplementaria 2). Tres de los 5 casos de TP53(P72R)-positivos mostraron niveles elevados de LDH y síntomas sistémicos.
Las mutaciones probablemente patogénicas en genes relacionados con la epigenética (TET2, DNMT3A, EZH2, ARID1A, ARID1B, NCOR) solo se encontraron en el 30% de los pacientes con SPTCL (casos n.° 6, 8 y 10). Estos 3 pacientes tenían la misma variante Y735C en DNMT3A. El cambio se encontraba en el dominio de la metiltransferasa del gen (fig. 2).
La pirosecuenciación reveló que solo un caso (caso 35) —una mujer de 26 años con síntomas sistémicos sin HPS— presentaba la mutación HAVCR2 (Y82C) en homocigosis (fig. 3).

Ninguno de los casos de LEP presentaba ninguna de estas mutaciones, incluida la variante TP53(P72R).
DiscusiónLos datos aquí presentados sugieren la presencia de diferencias etiopatogénicas entre el SPTCL y el LEP. Para poder comprender el fondo del LEP sería necesario la utilización de una tecnología con un mayor rendimiento. En cuanto al SPTCL, nuestros datos difieren de estudios anteriores.
La mutación de la línea germinal del receptor celular 2 del virus de la hepatitis A (HAVCR2, que codifica para la inmunoglobulina de células T y la proteína 3 que contiene el dominio de la mucina [TIM-3]) parece ser el sello distintivo del SPTCL7. En estudios previos se ha informado acerca de un elevado número de pacientes con SPTCL esporádico, oscilando entre el 25 y el 85% de los casos analizados6,7,12,13. El porcentaje más bajo de casos positivos se comunicó en la serie europea de Sonigo et al.12. Se pudieron analizar trece de los 53 casos, de los cuales solo 3 eran de origen europeo (es decir, el 23% de los casos positivos y el 6% de toda la serie). Solo un paciente (10%, 1/10) de nuestra serie, y 0 de los 10 casos estudiados con LEP presentaban SPTCL mutado. Estos datos sugieren un trasfondo diferente para los pacientes asiáticos y europeos con respecto al desarrollo del SPTCL. El impacto de los antecedentes genéticos, y el de los factores microambientales, en la distribución mundial de otros subgrupos del SPTCL se ha descrito anteriormente14–16.
Se han descrito 3 mutaciones principales en el gen HAVCR2 estrechamente relacionadas con el origen étnico de los pacientes: las variantes Y82C y T101I, que se asocian con la ascendencia asiática y polinesia, y la I97M, que se ha encontrado con mayor frecuencia en pacientes de origen norteafricano o caucásico.
También debemos mencionar que la variante I97M solo se ha descrito muy raramente en poblaciones asiáticas7, mientras que la variante Y82C solo se ha encontrado ocasionalmente en poblaciones distintas de la asiática, en la mayoría de los casos en heterocigosis6,12. De hecho, solo se ha encontrado un caso de la variante Y82C de la mutación del gen HAVCR2 en homocigosis en un paciente sudamericano6. Curiosamente, nosotros encontramos la variante Y82C en homocigosis en una mujer española de 26 años sin antecedentes familiares extranjeros conocidos ni antecedentes familiares previos de linfoma o enfermedad autoinmune. Los pacientes con mutaciones en HAVCR2 tienen una mediana de edad más joven al inicio de la enfermedad, predominio del sexo masculino, una mediana de tiempo más larga hasta el diagnóstico, un curso más grave de la enfermedad, una mayor tasa de autoanticuerpos y más probabilidades de enfermedad sistémica y desarrollo de HPS6,13,17,18. Además, los tumores con mutaciones en HAVCR2 son ricos en vías de señalización inflamatoria, IL6-JAK-STAT3, TNF-α y NFK-B. Por el contrario, los casos no mutados destacan por un perfil autoinmune y de «homing» linfocitario. Nuestra paciente con HAVCR2-mutado presentaba síntomas sistémicos, pero no HPS. De hecho, ninguno de los pacientes estudiados en esta serie presentaba HPS.
Las diferencias respecto a estudios anteriores pueden estar asociadas a diferencias de etnia, edad de inicio de la enfermedad, antecedentes familiares de linfoma y porcentaje de casos con SPTCL en la presente serie.
Hemos notificado, por primera vez, la presencia de la mutación del gen TP53(P72R) en los SPTCL. Se ha informado de que esta mutación es un polimorfismo de un solo nucleótido (SNP) común, con un sesgo étnico significativo, por lo que está presente en homocigosis en hasta el 40% de los estadounidenses caucásicos frente a solo ∼8% de los afroamericanos. Sin embargo, no estaba presente en ninguno de los 10 LEP analizados. Aunque se supone que este SNP no influye en el riesgo de cáncer, se ha asociado con un mayor peso, riesgo de diabetes tipo 2 e inflamación19. Esta propiedad inflamatoria se ha implicado en el aumento de varios subtipos de agresividad tumoral20.
Se encontraron diferencias fenotípicas entre los casos de SPTCL positivos y negativos para la variante TP53(P72R). Los casos positivos para la variante TP53(P72R) presentaban una regulación al alza y a la baja de las vías Treg/CD28 y metabólica, respectivamente. Se ha descrito una estrecha relación entre el CD28, el TNF y el desarrollo de Treg21. Se ha descrito un aumento del TNF y una menor actividad metabólica en ratones transgénicos R7219. Además, se ha demostrado que el p53 es un importante modulador de la diferenciación de células T CD4+, incluidas las células Th17 y Treg22. No se encontraron diferencias en la expresión de la proteína p53 entre los casos positivos y negativos para la variante TP53(P72R). Además, se ha propuesto que las células Treg son los «actores clave» en la interacción entre el metabolismo y la inmunidad. Se ha descrito una correlación inversa entre los niveles de leptina (una adipocitoquina producida por el tejido adiposo en respuesta a la cantidad de grasa) y la abundancia de células Treg en enfermedades autoinmunes23. Se han descrito niveles elevados de leptina en el lupus eritematoso sistémico (LES)24, y un metaanálisis ha descrito que la actividad del p53 no parece correlacionarse con la patogénesis de dicha enfermedad24.
Curiosamente, las mutaciones detectadas aquí podrían ser dianas útiles en relación con el tratamiento. Existen varias estrategias dirigidas contra el TP53 mutante oncogénico en los cánceres. El polimorfismo en el codón 72 (Arg/Pro) del TP53, el factor de transcripción codificado por el TP53, afecta a la sensibilidad celular a fármacos anticancerígenos como la doxorrubicina a través de la inhibición del p73, una proteína relacionada con el p5325. Además, los fármacos dirigidos a cambios epigenéticos han mostrado resultados prometedores26–28. Romidepsina (un inhibidor de HDAC)8 ha demostrado recientemente ser una terapia eficaz para el linfoma subcutáneo de células T similar a la paniculitis, dando una respuesta completa como monoterapia en casos resistentes al tratamiento de la enfermedad29.
Nuestro estudio amplía el panorama mutacional del SPTCL, sugiriendo que existe un importante trasfondo étnico subyacente al desarrollo de esta enfermedad, y resaltando la relevancia de los diferentes trasfondos moleculares en el desarrollo del SPTCL y el LEP. Sin embargo, nuestra serie es limitada y se necesitan más estudios en una serie de casos más amplia para validar nuestros hallazgos.
Este trabajo se ha realizado gracias al apoyo de becas del Instituto de Salud Carlos III (ISCIII) del Ministerio de Economía y Competitividad de España, cofinanciadas por la Unión Europea (FEDER) (FSE/FEDER, «Una manera de hacer Europa»/«Invertir en tu futuro» [MINECO, ISCIII]), financiado por la Unión Europea Next Generation EU, fondos que respaldan las acciones del Mecanismo de Recuperación y Resiliencia (MRR) (Plan Nacional I+D+I: PI17/02172, PI21/01724, 15826/004, 41163/005 y PMP21/00015), AECC, la Comunidad de Madrid y STARTUP2020/L2566. R.A.-A. ha recibido una beca predoctoral PFIS; L.T.-R. recibe una beca individual Marie Skłodowska-Curie (n.° 882597); M.R.-M. cuenta con el apoyo de CIBERONC (CB16/12/00291); P.M. tiene un contrato Miguel Servet financiado por el ISCIII (CP16/00116) y L.d.l.F. cuenta con el apoyo del contrato CA18/00017 del ISCIII.


