




El diagnóstico de la neurofibromatosis tipo 1 (NF1) habitualmente se demora en niños sin antecedentes familiares. Nuestro objetivo fue definir la prevalencia y características de las manifestaciones cutáneas prevalentes en la NF1, en comparación con la población general, que siguen siendo excluidas de los criterios diagnósticos para NF1.
Pacientes y métodosEstudio de casos y controles, pareado por grupos de edad, en el que se incluyó a 108 pacientes diagnosticados de NF1 y 137 controles sanos.
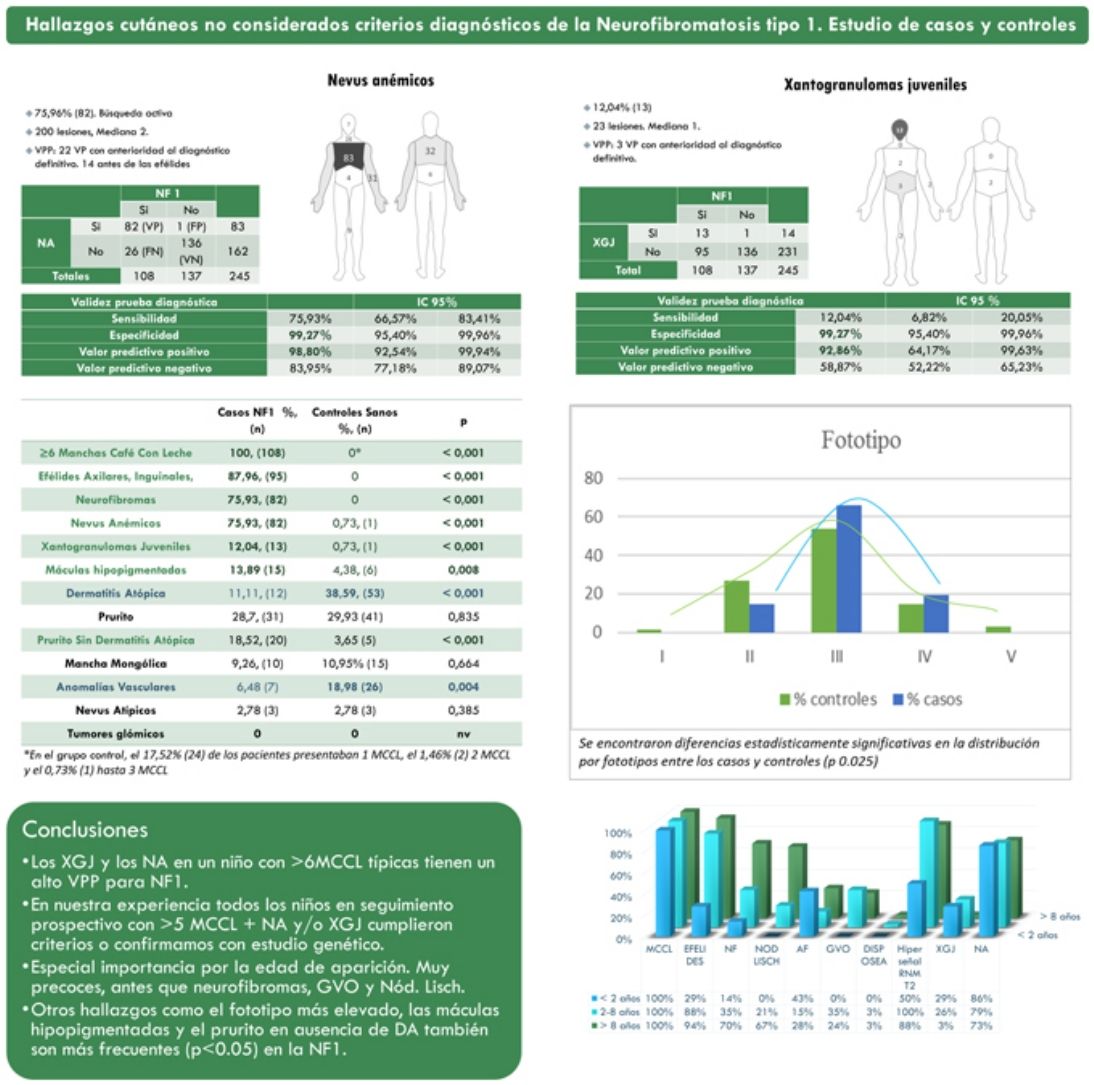
ResultadosLa prevalencia de nevus anemicus (NA) (p < 0,001) y xantogranuloma juvenil (XJ) (p < 0,001) fue significativamente superior en la población afectada de NF1, en comparación con el grupo control. Se estimaron una especificidad del 99,27% (intervalo de confianza: 95,4-99,96%) y un valor predictivo positivo (VPP) del 98,80% (92,54-99,94%) para NA, y una especificidad del 99,27% (95,4-99,96%) y VPP del 92,86% (64,17-99,63%) para XJ en el diagnóstico de NF1 en niños que presentan 6 o más manchas café con leche. También se evidenciaron diferencias estadísticamente significativas en la distribución por fototipos (p = 0,025) y con el generalizado sin otra causa conocida (p <,001).
ConclusionesLos NA y los XJ son hallazgos clínicos relevantes para el diagnóstico de NF1, especialmente durante los primeros años de vida. Consideramos que debería evaluarse su inclusión en los criterios diagnósticos de la enfermedad.
The diagnosis of Neurofibromatosis type 1 (NF1) is usually delayed in children without a family history. We aimed to define the prevalence and characteristics of prevalent skin manifestations in NF1 compared to the general population, which continue to be excluded from the diagnostic criteria for NF1.
Patients and methodsCase–control study, matched by age groups, in which 108 patients with a diagnosis of NF1 and 137 healthy controls were included.
ResultsThe prevalence of nevus anemicus (NA) (P<.001) and juvenile xanthogranulomas (JXG) (P<.001) was significantly higher in the population affected by NF1 than in the control population. A specificity of 99.27% (confidence interval): 95.4–99.96%] and a positive predictive value (PPV) of 98.80% [92.54–99.94%] were estimated for NA and a specificity of 99.27% [95.4–99.96%] and a PPV of 92.86% [64.17–99.63%] for JXG in the diagnosis of NF1 in children who present 6 or more Café-au-lait macules. Statistically significant differences were also evidenced in the distribution by phototypes (P=.025) and in relation to generalized itching with no other cause (P<.001).
ConclusionsNA and JXG are relevant clinical findings for the diagnosis of NF1, especially during the first years of life. We consider that its inclusion among the diagnostic criteria of the disease should be evaluated.
La neurofibromatosis tipo 1 (NF1; OMIM 613113, enfermedad 162200) constituye uno de los trastornos neurocutáneos hereditarios más comunes. El diagnóstico de la NF1 no plantea dificultades en los adultos. Sin embargo, en niños, y en especial en aquellos sin antecedentes familiares, el diagnóstico se demora varios años1. La inclusión de los nevus anemicus (NA) y del xantulogranuloma juvenil (XJ) se ha propuesto repetidamente entre las manifestaciones cutáneas que se consideran criterios diagnósticos para la NF1 en niños, basándose en diversos estudios observacionales retrospectivos y prospectivos2-5. A pesar de ello, la revisión reciente de los criterios diagnósticos de los National Institutes of Health (NIH) basados en un proceso Delphi modificado no logró el consenso (puntuación media 4-6/10) para incluir a los NA entre los nuevos criterios diagnósticos6. Los autores aluden a la falta de sensibilidad y especificidad, así como a la dificultad de su identificación por parte de facultativos no dermatólogos. El XJ no fue incluido posiblemente a causa de su naturaleza transitoria, a pesar de la existencia de consenso. Paradójicamente estos autores sí consideraron la inclusión de alteraciones coroideas observadas mediante tomografía de coherencia óptica/imagen de reflectancia en el infrarrojo cercano.
En este estudio, nuestro objetivo fue definir el valor predictivo positivo (VPP) y la fortaleza de la asociación entre las manifestaciones cutáneas más frecuentes en la NF1 que siguen sin considerarse criterios diagnósticos (fig. 1), a través de un estudio de casos y controles. De igual modo, tratamos de identificar las asociaciones con otras manifestaciones cutáneas que observamos a menudo entre los niños con NF1 y que han recibido menos atención en la literatura, tales como el prurito generalizado, la hiperpigmentación difusa o las máculas hipopigmentadas (MH)7.
Material y métodos Se observan 2XJ en la región facial de una niña de 2 años que solo presentaba MCCL y efélides. Las pruebas genéticas confirmaron el diagnóstico de NF1. b) Puede observarse un NA moteado mayor de 10 cm en el tórax y el cuello de un varón de 14 años.")
Se utilizó un diseño transversal de estudio de casos y controles pareado por grupos etarios, incluyendo exclusivamente a aquellos pacientes con diagnóstico confirmado de NF1. Los grupos de edad se establecieron conforme a la edad de inicio de los principales eventos diagnósticos (menor de 2 años, entre 2 y 8 años, y mayor de 8 años). Para el grupo control, se estimó que la muestra debía incluir al menos 137 niños sanos, con un nivel de confianza del 95%, y una potencia estadística del 80%, esperando encontrar diferencias de ± 8 puntos porcentuales.
Se incluyó como casos a todos los pacientes menores de 18 años de edad con diagnóstico definitivo de NF1 (cumpliendo más de 2 criterios, o 2 criterios y la detección de mutaciones del gen de NF1 para aquellos que solo presentaron manchas café con leche (MCCL) y efélides en los pliegues), que acudieron a nuestra unidad multidisciplinar de NF1 entre el 1 de mayo del 2012 y el 10 de febrero del 2017, tras haber obtenido el consentimiento informado de sus tutores. En cuanto a los controles sanos, se incluyó a los niños menores de 18 años de edad, sus pacientes o acompañantes sanos examinados entre el 1 de octubre del 2016 y el 10 de febrero del 2017, cuyo motivo de consulta no coincidiera con ninguna de las variables principales del estudio.
Durante las visitas, y tras revisar sus historias clínicas, se recopilaron los datos relativos a la historia familiar, la historia dermatológica personal, el fototipo y los hallazgos clínicos del examen, relacionados o no con la NF1.
También se contrastaron las variables referentes a las manifestaciones cutáneas más relevantes para NF1 con el resto de hallazgos dermatológicos apreciados durante el estudio, las principales comorbilidades asociadas a la NF1 y los criterios diagnósticos de la NF1. Se utilizó el contraste χ2 de Pearson o el test exacto de Fisher, según la aplicabilidad o no del primero. Todos los contrastes se realizaron a dos colas y se consideraron significativos los valores de p menores del 5%. El programa estadístico utilizado fue STATA/SE versión 10.0 (StataCorp) y Excel (Microsoft Office). Para las variables que reflejaron asociaciones estadísticamente significativas, se calculó la dirección de la asociación utilizando odds ratio (material complementario en el Anexo B).
El protocolo del estudio fue revisado y aprobado por el comité de ética e investigación del Hospital Niño Jesús de Madrid, R-0024/16 Acta n° 11/16, 27 de septiembre de 2016.
ResultadosDe una cohorte de 135 niños bajo seguimiento de NF1 por parte del Servicio de Dermatología de nuestro centro, se incluyó a 108 niños con diagnóstico confirmado de NF1 y 137 controles sanos de edades comprendidas entre 2 meses y 18 años. La prevalencia de los NA (p<0,001) y XJ (p<0,001) fue significativamente superior en la población afectada de NF1 en comparación con la población control. En 82 de los casos se objetivaron NA, mientras que en los controles se evidenció un único NA. Estimamos por tanto que los NA tienen una sensibilidad 75,9% (66,6-83,4%), una especificidad del 99,3% (95,4-99,9%), un VPP del 98,8% (92,5-99,9%) y un valor predictivo negativo (VPN) del 84% (92,5-99,9%) para el diagnóstico de NF1. En el caso de los XJ, la sensibilidad fue del 12% (6,8-20,1%), la especificidad del 99,3% (95,4-99,9%), el VPP del 92,9% (64,2-99,6%) y el VPN del 58,9% (52,2-65,2%). La tabla 1 resume las características clínicas de NA y XJ.
Características clínicas de nevus anemicus y xantogranuloma juvenil
| Signos clínicos | Nevus anemicusn (%) | Xantogranuloma juveniln (%) |
|---|---|---|
| 108 pacientes | 82 (76) | 13 (12) |
| Sexo | p=0,124 | p=0,026 |
| Varón | 45 (54,9) | 4 (30,8) |
| Mujer | 37 (45,1) | 9 (69,2) |
| Frecuencias | ||
| Recuento de lesiones | 200 | 23 |
| Media | 2,4 (DE±1,8) | 1,8 (DE±1,2) |
| Mediana [rango] | 2 [1-12] | 1 [1-4] |
| Localización | ||
| Cabeza y cuello | 35 (17,5) | 12 (52,2) |
| Torácica | 115 (57,5) | 2 (8,7) |
| Abdominal | 4 (2) | 3 (13) |
| Lumbar | 6 (3) | 2 (8,7) |
| Brazos | 31 (15,5) | 2 (8,7) |
| Miembros inferiores | 9 (4,5) | 2 (8,7) |
| Tamaño | ||
| <1cm | 91 (45) | 22 (95,6) |
| 1-5cm | 81 (41) | 1 (4,4) |
| > 5cm | 28 (14) | 0 |
| Morfología | ||
| Redondeado u oval | 94 (47) | 23 (100) |
| Lobulado | 53 (26) | 0 |
| Moteado | 50 (25) | 0 |
| Agrupado | 3 (2) | 0 |
DE: desviación estándar.
La distribución por grupos de edad de los criterios diagnósticos y los hallazgos más relevantes se muestra en la figura 2. Durante el periodo del estudio, incluimos 22 pacientes en los que objetivamos la presencia de NA con anterioridad al establecimiento del diagnóstico definitivo. En el 63,6% de ellos (14), los NA se constataron antes de observar las efélides axilares o inguinales. En 7 de ellos la demostración posterior de nódulos de Lisch (3) o de neurofibromas (4) permitió verificar el diagnóstico. En los 15 casos restantes, se confirmó el diagnóstico mediante estudio genético antes de demostrar la presencia de otros criterios diagnósticos. Como en el caso de NA, pudimos observar 3 pacientes con XJ antes de que presentaran criterios diagnósticos de NF1.

La tabla 2 muestra los resultados del estudio de casos y controles relativos a otras manifestaciones dermatológicas. Se observaron diferencias estadísticamente significativas en la distribución por fototipos (p=0,025), presentando los pacientes con NF1 un fototipo proporcionalmente más alto (III-IV) al de la población control (fig. 3). Consideramos relevante que se objetivaran señales hiperintensas en la resonancia magnética nuclear (RMN) cerebral en el 91,1% de los pacientes incluidos en este estudio. Los resultados más relevantes del análisis de las variables se recopilan en el material complementario incluido en el Anexo B.
Resultados del estudio de casos y controles y significación estadística
| Casos de NF1n (%) | Controles sanosn (%) | p | |
|---|---|---|---|
| Manifestaciones cutáneas consideradas criterios diagnósticos por los NIH | |||
| ≥ 6 MCCL | 108 (100) | 0a | < 0,001 |
| Efélides axilares, inguinales | 95 (88) | 0 | < 0,001 |
| Neurofibromas | 82 (75.9) | 0 | < 0,001 |
| Manifestaciones no consideradas criterios diagnósticos por los NIH | |||
| Nevus anemicus | 82 (75,9) | 1 (0,7) | < 0,001 |
| Xantogranuloma juvenil | 13 (12)) | 1 (0,7) | < 0,001 |
| Máculas hipopigmentadas7 | 15 (13,9) | 6 (4,4) | 0,008 |
| Dermatitis atópica | 12 (11,1) | 53 (38,6) | < 0,001 |
| Prurito | 31 (28,7) | 41 (29,9) | 0,835 |
| Prurito sin dermatitis atópica | 20 (18,5) | 5 (3,7) | < 0,001 |
| Melanocitosis dérmica | 10 (9,3) | 15 (11) | 0,664 |
| Anomalías vasculares | 7 (6,5) | 26 (19) | 0,004 |
| Nevus melanocíticos atípicos | 3 (2,8) | 3 (2,8) | 0,385 |
Los valores en negrita representaron las variables con diferencias estadísticamente significativas (p < 0,05).

La observación de 6 o más MCCL redondeadas u ovaladas, mayores a 5 mm, en un lactante es altamente sugestiva y obliga a descartar en primer lugar la NF18, aunque dichos hallazgos no son patognomónicos9-12. La descripción de otros síndromes que pueden manifestar también MCCL y efélides durante los primeros años de vida, tales como el síndrome de Legius, síndrome constitutivo de deficiencia de reparación de emparejamientos erróneos ha puesto de manifiesto que por sí solos los criterios pigmentarios son insuficientes para considerar el diagnóstico de la NF1 probado. Esta circunstancia nos obliga a decidir entre confirmar nuestra sospecha diagnóstica mediante el estudio genético o esperar al desarrollo de otros criterios manteniendo indefinidamente la ansiedad que provoca este diagnóstico entre los progenitores9. Por este motivo, con anterioridad a la publicación de los criterios revisados de Legius et al.6, incluimos en nuestra muestra exclusivamente a los pacientes con diagnóstico confirmado de NF1, para obtener conclusiones con mayor significación estadística. Se excluyeron, por lo tanto, 27 pacientes que solamente presentaran MCCL y efélides y también a aquellos pacientes con sospecha diagnóstica de NF1 mosaico. En consecuencia, el porcentaje de pacientes de nuestra serie que presentaron al menos 3 de los criterios diagnósticos superó el 81%. En el resto de los pacientes, que exclusivamente presentaron criterios diagnósticos pigmentarios, es decir, MCCL y efélides, se confirmó el diagnóstico mediante estudio genético, siendo por tanto, muy poco probable la existencia de falsos negativos en nuestra serie.
En la última década, se ha evidenciado que el NA representa una manifestación característica de la NF1. Al tratarse de un estudio prospectivo y fruto de la experiencia acumulada en la búsqueda y reconocimiento de estas lesiones, la prevalencia de NA en nuestra serie actual asciende al 76%, superando incluso nuestro propios resultados publicados en 20153. Se demuestra por tanto, la importancia de la búsqueda activa de los NA, puesto que pueden pasar desapercibidos en el examen físico de rutina (fig. 1b). Para detectarlos, recomendamos preguntar rutinariamente a los padres si han visto lesiones blanquecinas transitorias durante el baño, los procesos febriles, el llanto o el ejercicio, y frotar enérgicamente el área preesternal y la espalda de los bebés y niños pequeños para resaltar las lesiones. Aunque han sido consideradas lesiones congénitas, la edad de aparición de estas no se conoce con exactitud. En nuestra serie, se identificaron numerosos NA en niños menores de 2 años. Las principales publicaciones sobre NA en la NF1 hacen referencia a la población pediátrica3-5, por lo que desconocemos la prevalencia real de los NA en pacientes adultos. Algunos autores han sugerido que el NA puede quedar camuflado por el desarrollo de neurofibromas13. También se ha descrito la regresión de NA7, aunque en nuestra serie encontramos únicamente un caso de regresión. Nuestros propios datos evidencian que la prevalencia de los NA es proporcionalmente mayor en los pacientes incluidos en el grupo etario de menores de 2 años (86%) respecto al de los mayores de 8 años (73%). Esta circunstancia es un hecho contrastado en otros rasgos clínicos de la NF1 como en los gliomas de la vía óptica (GVO) y las señales hiperintensas en la RNM14. La fisiopatogenia de NA no ha sido confirmada hasta el momento. La localización, morfología y tamaño no serán objeto de discusión puesto que los resultados no difieren de los publicados en 20153.
Para nuestro conocimiento, actualmente se ha descrito únicamente un caso de NA relacionado con el síndrome de Legius15 y otro con el complejo de esclerosis tuberosa (CET)16. Por tanto, son necesarios nuevos estudios prospectivos sobre otras rasopatías, para esclarecer si los NA constituyen hallazgos específicos de NF1, como sospechamos. Algunos autores mantienen que la detección de NA en los pacientes con MCCL o efélides confirmarían el diagnóstico de NF117. Nuestros resultados demuestran que los 22 niños afectos por los criterios pigmentarios cutáneos y NA fueron posteriormente diagnosticados de NF1 (VPP teórico del 100%) y que la presencia de NA podrían haber anticipado el diagnóstico en caso de considerarlos como criterio, incluso antes de la aparición de las efélides axilares o inguinales. Además, la edad temprana de aparición de NA facilitaría el diagnóstico precoz de la enfermedad. De igual modo, al observar uno o múltiples NA en un niño, recomendamos investigar en el mismo otros signos de la NF1.
Aunque menos prevalente que el NA, los XJ son considerados lesiones frecuentes en los niños afectados de NF1. La frecuencia de XJ en los pacientes pediátricos varía considerablemente en las diferentes publicaciones sobre NF1, oscilando entre el 3,9 y el 37,5% de los niños18,19. Nuestros resultados reflejaron una prevalencia del 12% aunque, sin embargo, al finalizar el estudio dicha prevalencia descendería al 5,6% debido a la naturaleza transitoria del XJ. En nuestra serie, el 80% de los XJ fueron registrados durante los primeros 2 años de vida, desapareciendo al cumplir 1 o 2 años, y siendo poco frecuentes en los pacientes mayores de 5 años (fig. 1a). Tampoco se reflejarán nuestros resultados en cuanto a localización, morfología y tamaño, ya que coinciden con los datos previamente publicados20. Destacaríamos la existencia de una diferencia estadísticamente significativa en la distribución por sexos, siendo predominante la presencia de los XGJ en niñas. Este hecho no se ha reflejado previamente en la literatura.
La NF1 es un trastorno caracterizado por hiperpigmentación, a pesar de lo cual subrayamos la elevada frecuencia de MHs registradas en nuestra serie (13,9%) (tabla 2). Justificamos esta marcada prevalencia por la búsqueda activa de NA, que nos ha permitido identificar estas lesiones, a veces sutiles, y que no se acentúan mediante la fricción, a diferencia de los NA. Estas MH podrían considerarse nevus acrómicos o nevus despigmentados pero, dada la ausencia de estudios anatomopatológicos o moleculares y su similitud con las lesiones descritas por Riccardi en 198721, decidimos conservar la denominación tradicional7. Las similitudes clínicas de las MHs de morfología ovalada con las manchas en hoja de fresno o lanceoladas del complejo esclerosis tuberosa (CET) resultan evidentes. Aunque de manera excepcional, se ha publicado con anterioridad la asociación de NF1 y TSC en el mismo paciente22. Destacamos que ninguno de nuestros pacientes con máculas hipocrómicas, ni sus familiares, presentaron otros signos indicativos de CET. En el grupo control, observamos la presencia de MH en un porcentaje considerablemente alto (4,4%) y, a pesar de ello, confirmamos la existencia de una asociación estadísticamente significativa entre MH y NF1 (p=0,008). Antes de establecer la relación causal entre ambos procesos, son necesarios estudios tisulares y moleculares para identificar el mecanismo patogénico que da lugar a dicha hipopigmentación localizada.
En referencia al resto de hallazgos dermatológicos que en la actualidad carecen de transcendencia en el diagnóstico de la enfermedad, nos gustaría resaltar las diferencias estadísticamente significativas referentes al fototipo y al prurito generalizado en pacientes sin antecedentes u otros signos clínicos de dermatitis atópica (DA) entre los pacientes y los controles. Este resultado nos ha permitido corroborar las observaciones de los propios pacientes y de los autores que aseveraban que los pacientes con NF1 presentan fototipos más oscuros23 y que padecen de prurito generalizado con mayor frecuencia que la población control24. Por el contrario, no hemos sido capaces de confirmar la repercusión diagnóstica de los tumores glómicos en la NF1, ya que no los hemos observado en ninguno de los 2grupos. Su baja frecuencia en los pacientes pediátricos reduce su papel como criterio diagnóstico, aunque su detección motivará la búsqueda de otras características de NF1 en los pacientes adultos25.
Consideramos que la prevalencia de otras patologías cutáneas como anomalías vasculares o la DA fue significativamente más elevada en el grupo de controles debido a un sesgo de selección, puesto que son motivos de consulta muy frecuentes en dermatología pediátrica.
Coincidimos con Legius et al.6 que resulta esencial reconocer las limitaciones, reinterpretar y adaptar el diagnóstico de la NF1 basado en los criterios diagnósticos establecidos en 1987 por los NIH. Este estudio de casos y controles demuestra que los NA y XJ son hallazgos más prevalentes en los pacientes con NF1 que en la probación general, y confirma la elevada especificidad y VPP de NA y XJ para el diagnóstico de NF1. De igual modo, consideramos la utilidad diagnóstica de estas manifestaciones cutáneas probadas, dado que nos permitirán confirmar este diagnóstico a una edad más temprana, sin las repercusiones económicas y sanitarias asociadas al estudio molecular, o las pruebas complementarias recientemente propuestas.
Por tanto, este estudio respalda con suficiente evidencia la inclusión de estos 2signos como criterios diagnósticos para NF1 o, al menos indican la realización de pruebas genéticas cuando observamos NA o XJ en niños con 6 o más MCCL.
Otros hallazgos como el fototipo más elevado, las MH y el prurito generalizado también son hallazgos prevalentes en los niños que padecen NF1, que deben tenerse en cuenta en el diagnóstico clínico.
Una vez analizados nuestros resultados, consideramos probado que la exploración clínica minuciosa nos permitirá diagnosticar la NF1 con mayor precisión y a edades más tempranas.
FinanciaciónNo se ha recibido financiación para la realización de este trabajo. Este trabajo está incluido en la tesis doctoral distinguida con la beca titulada Áccesit a la mejor Tesis Doctoral Edición 2017 de la «Sección Centro» de la Academia Española de Dermatología y Venereología-SC-AEDV.
Presentaciones previasEste estudio se incluye en la Tesis Doctoral titulada: «Estudio clínico ecográfico de la Neurofibromatosis Tipo 1 en la edad pediátrica» (https://repositorio.uam.es/bitstream/handle/10486/680203/garcia_martinez_francisco_javier.pdf?sequence=1&isAllowed=y).
Los resultados y conclusiones incluidos en esta tesis doctoral han sido previamente publicados o aceptados para su publicación:
García-Martínez F.J., et al. Congenital cutaneous neurofibromas in neurofibromatosis type 1: Clinicopathological features in early infancy. J Dtsch Dermatol Ges. 2021 Jan;19(1):73-80. doi: 10.1111/ddg.14322. Epub 2021 Jan 14. PMID: 33448128.
García-Martínez F.J., et al. Hypopigmented macules in neurofibromatosis type 1: A case control study. J Am Acad Dermatol. 2021 Apr;84(4):1128-1130. doi: 10.1016/j.jaad.2020.06.071. Epub 2020 Jun 24. PMID: 32592882.
García-Martínez F.J., et al. Clinical and sonographic classification of neurofibromas in children with NF1. A cluster analysis. Ultraschall in der Medizin/European Journal of Ultrasound. 2021 Nov 24. doi: 10.1055/a-1640-9621.
Declaración de consentimientoTodos los participantes y/o sus tutores legales han acordado voluntariamente su participación en este estudio de investigación.
Conflicto de interesesLos autores declaran la ausencia de conflicto de intereses.
Datos complementariosLos datos complementarios asociados a este artículo pueden encontrarse en la versión online, en doi:10.1016/j.ad.2022.05.014.
A todos los médicos residentes que han colaborado en la recopilación de los datos.
Cómo citar este artículo: García-Martínez FJ, Duat-Rodríguez A, Andrés Esteban E, Torrelo A, Noguera Morel L and Hernández-Martín A, Cutaneous Manifestations not Considered Diagnostic Criteria for Neurofibromatosis Type 1. A Case-Control Study. https://doi.org/10.1016/j.ad.2022.05.014.


