







Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia, is a rare autosomal dominant disorder with an estimated worldwide prevalence of 1 case per 10,000 population. Its clinical manifestations are the result of arteriovenous malformations characterized by telangiectases that can affect the skin, mucous membranes, and solid organs and cause life-threatening conditions, such as liver disease, systemic emboli, and heart failure. Timely diagnosis is thus essential in order to prevent disease-related complications and offer genetic counseling to families. We review the clinical features of Osler-Weber-Rendu syndrome with a focus on mucocutaneous manifestations and their treatment.
El síndrome de Osler-Weber-Rendu, o síndrome hereditario hemorrágico telangiectasia, es un trastorno raro de herencia autosómica dominante con una prevalencia estimada de 1:10.000 personas a nivel mundial. Las manifestaciones clínicas de este síndrome son resultado de malformaciones arteriovenosas y varían desde telangiectasias en piel y mucosas hasta afección de órganos sólidos que ponen en peligro la vida, como alteraciones hepáticas, émbolos sistémicos y fallo cardíaco, por lo cual el diagnóstico oportuno es de suma importancia para prevenir las complicaciones de la enfermedad y proporcionar apoyo genético a los familiares. En esta revisión se analiza el cuadro clínico con enfoque principal en las manifestaciones mucocutáneas de la enfermedad y su abordaje terapéutico.
Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia (HHT), is a rare autosomal dominant disorder resulting from abnormal communication between veins and arteries. These arteriovenous malformations are manifest as telangiectases that can affect the skin, mucous membranes, and solid organs.1 The presence of skin telangiectases is a cosmetic problem for patients with HHT; however, systemic manifestations such as recurrent epistaxis, liver disease, systemic emboli, and heart failure can be life-threatening. The estimated worldwide prevalence is between 1:5000 and 1:10 000 population, but it is thought to be underdiagnosed.1,2 Patients with HHT require a multidisciplinary approach due to the different systemic complications that may be present. Physicians should therefore be familiar with this condition and contribute to timely diagnosis and treatment of the disease.
HistoryThe first recorded report of HHT was made by Sutton3 in 1864, when the author described a male patient with recurrent episodes of bleeding and vascular malformations. In 1865, Babington4 published a case of hereditary epistaxis in a boy and his mother, both of whom had recurrent episodes of epistaxis. Later, in 1896, Rendu5 was the first to describe the syndrome when he identified a man with clinical manifestations of facial telangiectases on the nose, tongue, and lips. This man also had a history of anemia and episodes of recurrent epistaxis from childhood onwards. His father reported episodes of melena. Subsequently, Sir William Osler6 was the first to report a family association in the syndrome when he described 3 families with telangiectases and hereditary bleeding. In 1907, Weber7 described the hereditary relationship between telangiectases and bleeding. In 1909, Hanes8 proposed the term HHT.
EpidemiologyHHT has a broad geographic distribution, and in recent years, given greater awareness and knowledge of the disease, the number of case reports has increased. The exact prevalence is therefore unknown, with estimates ranging from 1:2351 inhabitants in France and 1:3500 in Denmark to 1:39 216 in the North of England.9 Worldwide, the disease is estimated to affect 1.4 million people.
Although most patients have another family member with HHT, 20% of cases are sporadic. It is estimated that the offspring of someone with HHT has a 50% risk of developing the disease.10
PathophysiologyThe clinical manifestations observed in HHT are the result of abnormalities in vascular structure. The main lesions are telangiectases, which arise from arteriovenous shunting between dilated arterioles and venules and which present clinically as punctiform or linear arborizing erythematous macules of 1-2cm that disappear on applying light pressure.11 The pathophysiological mechanism by which telangiectases occur has not been fully elucidated. The Braveman theory proposes as an initial mechanism a focal dilatation of postcapillary venules, which continue to enlarge and eventually connect with dilated arterioles through capillaries that subsequently disappear to form a direct arteriovenous shunt. During this process, a predominantly mononuclear perivascular infiltrate of is present.12 By definition, telangiectases present in mucocutaneous tissues, such as the skin or gastrointestinal mucosa. When these abnormal vascular shunts occur in internal organs, such as the liver, lung, or brain, they are known as arteriovenous malformations (AVMs). The AVMs are responsible for the systemic manifestations of the disease.13 The fragility of the vascular walls and turbulent blood flow predispose these vessels to the characteristic bleeding that forms part of the syndrome.14
It has been observed that vascular endothelial growth factor (VEGF), which is implicated in vasculogenesis and angiogenesis, is elevated in patients with HHT, and so treatments such as bevacizumab that target this factor have been developed and shown to be effective.15
On the molecular level, more than 600 mutations causative of HHT have been identified. Of these, the most studied to date are mutations in the endoglin (ENG) gene, located on chromosome 9, characteristic of HHT subtype 1, and in the activin-like kinase (ALK1) gene, on chromosome 12, characteristic of subtype 2.11,16 Both the corresponding proteins (ENG and ALK1) are transmembrane proteins expressed in endothelial cells with the ability to bind to transforming growth factor (TGF). These molecules therefore play an important role in angiogenesis and vascular maturation.17 There have also been reports of mutations in the MADH4 and SMAD4 genes, which predispose the individual to HHT accompanied by juvenile polyposis (Table 1).11 Another type of mutation is one that affects bone morphogenic protein receptorII, leading to the HHT phenotype accompanied by primary pulmonary hypertension.18
Main HHT Mutations.
| ENG | Associated with mutations in the endoglin geneMore frequently observed in pulmonary and cerebral AVMs |
| ACVRL1 | Associated with mutations in the ALK-1 gene (activin-like kinase receptor)More frequently observed in hepatic AVMs, pulmonary and arterial hypertension |
| SMAD4 | Causes HHT-associated juvenile polyposis |
The clinical manifestations of HHT are varied, and present mainly on the nose, skin, and lungs, with possible involvement of the central nervous system and gastrointestinal system. They are characterized by the classic triad of epistaxis, telangiectases, and a family history of HHT.19
Most patients only experience manifestations of epistaxis, mucocutaneous telangiectases, and anemia due to iron deficiency. Symptoms are not normally present at birth but develop as the individual gets older. It is estimated that 70% of affected individuals present manifestations by 16 years and 90% by 40 years.9
NoseEpistaxis is usually the primary clinical manifestation, and in many cases, the first. Spontaneous bleeding occurs due to telangiectases that are present in the nasal mucosa. These events can be triggered my many factors, such as changes in temperature, humidity, or posture.
The frequency of nose bleeds can be very high, leading to iron-deficiency anemia. However, on other occasions, episodes may occur only sporadically or never, and so diagnostic suspicion is less likely. Furthermore, epistaxis is considered a very common symptom in the general population, and so it is not a very sensitive indicator. Episodes of night bleeding are more specific for diagnosis of HHT.20
Generally, episodes of epistaxis start in childhood, at around 10 years of age, and usually become more severe as the individual gets older. On average, patients with HHT present around 18 episodes of epistaxis a month. Rebeiz et al.21 developed a classification for severity of epistaxis that is still in use today (Table 2).
Management and prevention of epistaxis can vary from simple interventions, such as nasal saline spray, to more aggressive measures, such as cauterization, laser ablation, surgery, estrogen supplementation, and embolectomy, depending on the severity of the episodes.22,23
SkinThe main skin manifestations are telangiectases, which occur in up to 75% of patients, with onset typically in childhood and becoming more numerous with increasing age.
Skin lesions usually appear after the first episode of epistaxis. These lesions usually become apparent when patients are in their 30s and 40s.11
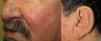
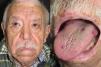
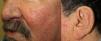
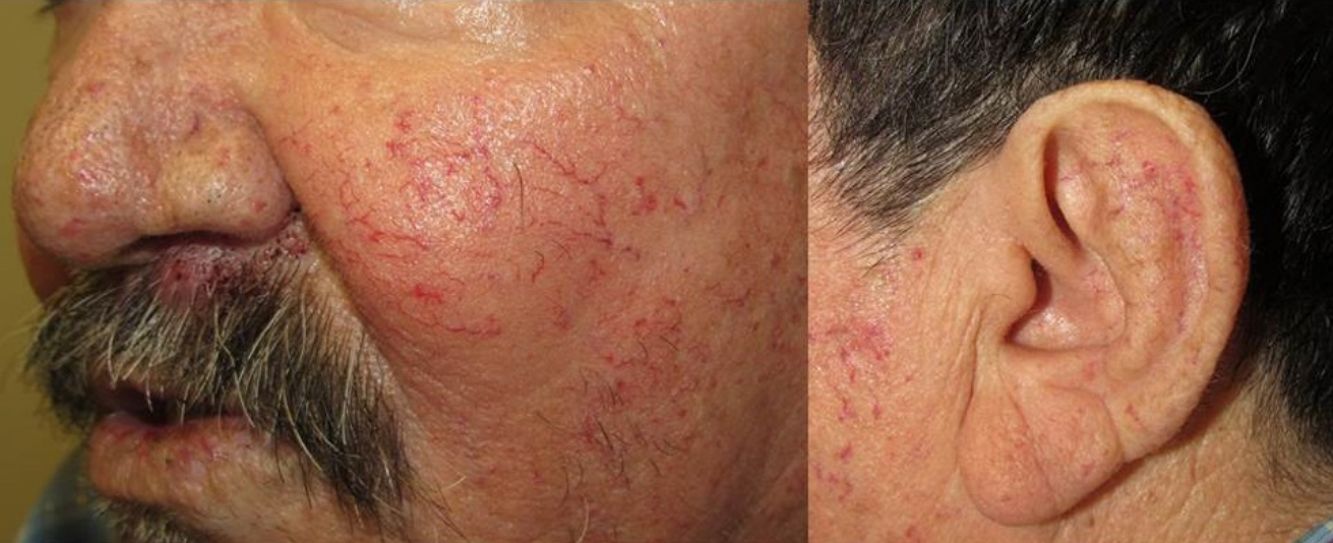
They present most frequently on the face, lips, nose, tongue, outer ears, hands (especially the fingertips), trunk, and feet (Figs. 1-3).19
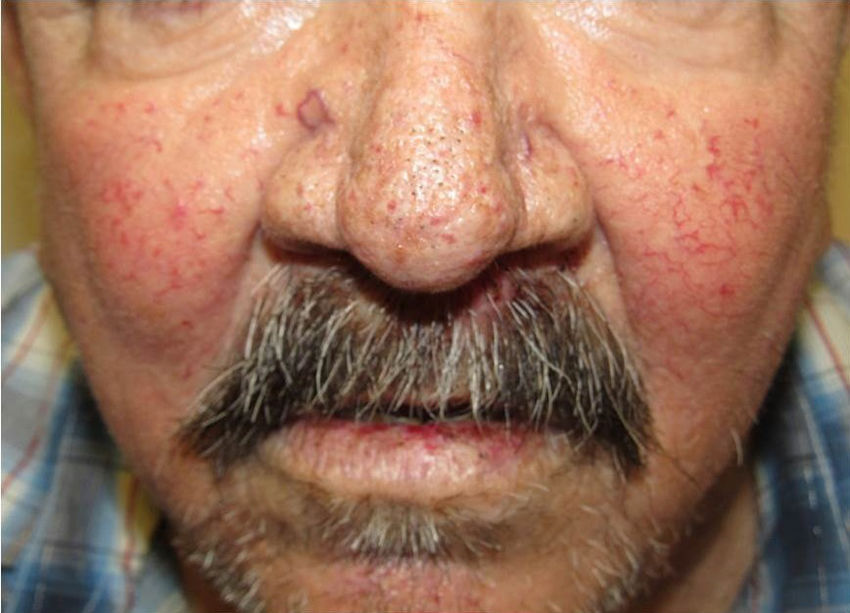
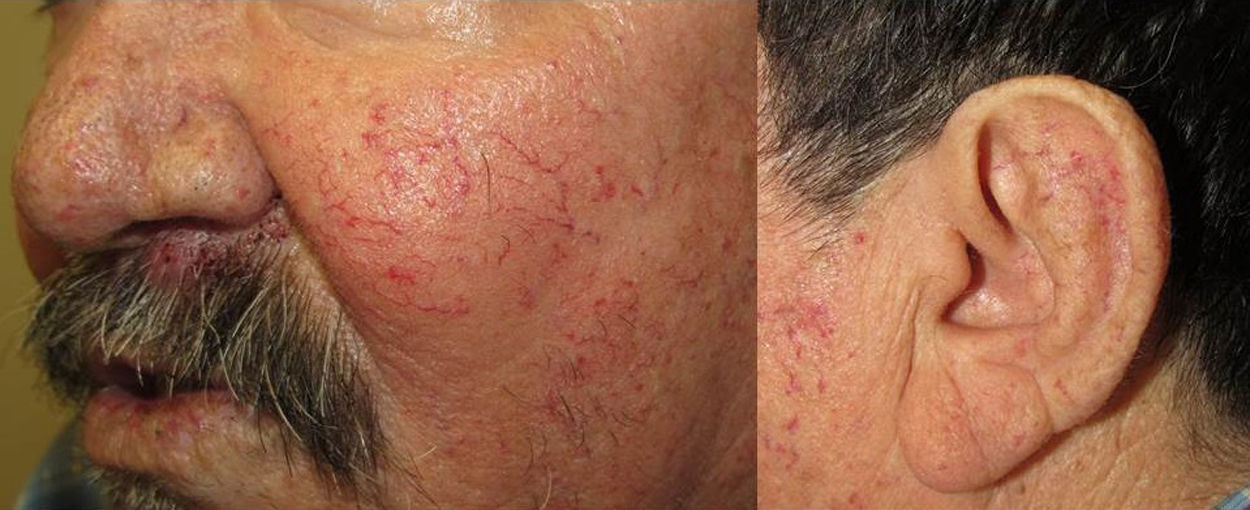
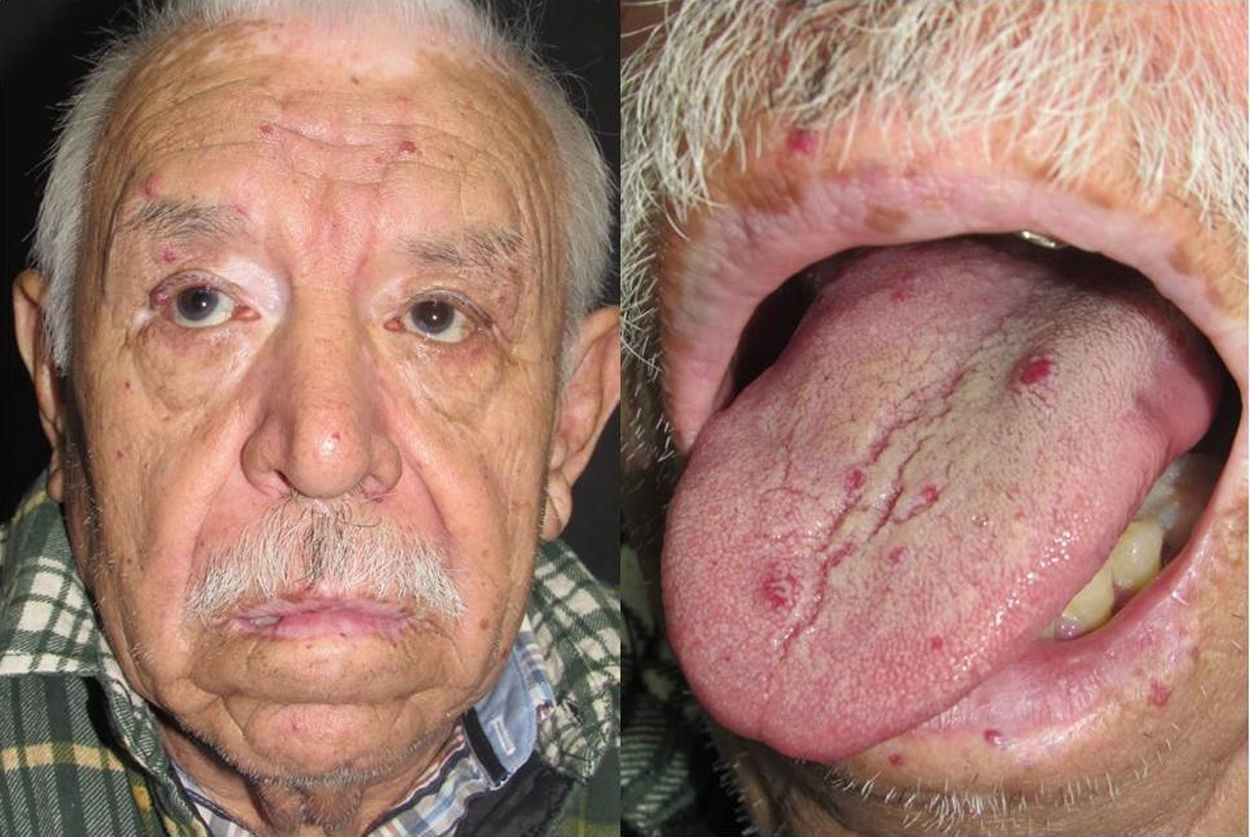
In the early stages, the lesions are small, pulsatile red-violaceous macules 1-3 in diameter, which in the chronic phase may coalesce and increase in diameter to form arborizing lesions.19,24
In most cases, cutaneous telangiectases are asymptomatic and treatment is a question of esthetics. However, in some cases, they can be painful.
The HHT Foundation International drew up a consensus in which they arrived at the conclusion that the presence of at least 3 typical telangiectases are required for diagnosis of HHT.17
LungThe most frequent pulmonary lesions are AVMs, which are present in up to 15% to 30% of patients with HHT. These AVMs directly connect pulmonary and systemic circulation, causing a right-to-left shunt that is manifest clinically as cyanosis, intolerance of cold, migraine, and polycythemia. The most serious complications include cerebrovascular accidents, massive hemoptysis, and spontaneous hemothorax.10
In general, these lesions occur in the lung bases, and bleed more readily during pregnancy.25
Given that pulmonary AVMs do not lead to symptoms in early stages and that complications may be very serious, all patients over 16 years of age with diagnosis of HHT should undergo transthoracic ultrasound. Lesions can be treated by transcatheter embolization or, in the most severe cases, lobectomy.10
Gastrointestinal EffectsGastrointestinal effects arise in a third of patients with HHT.
The most frequent lesions are telangiectases in the stomach and duodenum, followed by AVMs.
In the most usual clinical presentation, the patient will have iron-deficiency anemia or episodes of acute gastrointestinal bleeding; these manifestations are more common in individuals aged over 40 years.10 Gastrointestinal monitoring usually starts at the age of 35 years and involves only annual hemoglobin measurement; endoscopy is reserved for episodes of bleeding. Endoscopic ultrasound-guided ablation, embolization, or surgery are the treatments for gastrointestinal manifestations.
BrainBrain lesions present in 23% of patients. These lesions can be multiple AVMs, telangiectases, and arteriovenous fistulas. When present, they can manifest as episodes of migraine, convulsions, ischemia, or bleeding. The annual rate of AVM rupture is 2% to 4% on average. It is a matter of debate whether screening for early detection is appropriate, but if performed, magnetic resonance imaging is the technique of choice, with a sensitivity of 80% to 95%. Treatment can be by embolization or surgical resection.
LiverHepatic involvement in HHT is present in 32% to 78% of patients and symptoms are present in 8% of them. Clinical presentation can include angina and heart failure due to shunting between the hepatic artery and vein, which increases the cardiac overload. Portal hypertension and hepatic encephalopathy may also arise. These lesions can be monitored by Doppler ultrasound, magnetic resonance imagining, and computed tomography. Supportive therapy is administered, with the aim of reducing the complications of portal hypertension and heart failure. Sometimes, transplantation may be performed.14
DiagnosisTimely diagnosis of HHT is of utmost importance to prevent the complications of the disease and to provide genetic counseling for family members. In cases in which the classic 3 criteria of epistaxis, telangiectases, and a family history are present, diagnosis is relatively simple; however, not all patients present with this triad. In 2000, the Curaçao Diagnostic Criteria for HHT were published; 3 or more criteria are required for definitive diagnosis, 2 positive criteria correspond to a probable diagnosis, and no positive criteria are equivalent to unlikely diagnosis (Table 3).10,20
There are also genetic tests for diagnosis of HHT. The main use for these is for study of family members of affected patients, particularly children and young adults, who do not meet the clinical diagnostic criteria. Among these tests are those for DNA sequencing to detect mutations in the ENG and ACVRL1 genes, which account for the majority of mutations associated with HHT.26 There are also tests to detect mutations in the SMAD4 gene, but these are less common.27
Capillary MicroscopyDermoscopy has become a useful tool for the diagnosis of HHT through capillary microscopy. Capillary microscopy findings described to date in HHT suggest that the technique is useful because it can detect microscopic telangiectases. In a study conducted by Mager and Westermann,17 using capillary microscopy of the nail fold, the authors found that 87% of patients with HHT had vascular abnormalities, with giant loops being the most common finding.
TreatmentGiven that HHT syndrome has a multisystemic impact, multidisciplinary treatment is also required. Most studies of HHT are conducted by ear-nose-throat specialists because epistaxis is the main manifestation in these patients; however, although telangiectases are just as evident, few studies have been conducted of their treatment.
Although cutaneous telangiectases are only a cosmetic problem, their presence can have a negative impact on the quality of life of the patients, and so several treatment options have been implemented within dermatology, with varying results. Most articles on the subject are merely case reports.
In recent years, laser treatments have been the most widely used for treatment of cutaneous telangiectases. The most extensively used systems are pulsed dye lasers (595nm), which are useful for macular telangiectases, and Nd:YAG lasers, which are preferred for lesions with a papular appearance and because of their greater penetration and coagulation power (Table 4).28
Review of Case Reports of Telangiectases in HHT Treated With Laser Light.
| Study | Number of Patients | Treatment | Treated Areas | Outcome |
|---|---|---|---|---|
| Cheung et al.,28 2015 | 1 | Pulsed dye laser at 595nmSpot, 5mmPulse duration, 1.5 msFluence, 8J/cm2 on cheeks and 9J/cm2 on the nose | Face and mucosa | Resolution of lesions in 5 days |
| Halachmi et al.,29 2013 | 8 | Pulsed dye laser (Vbeam Perfecta, Candela, Wayland)Spot, 5-7mmPulse duration, 1.5 msFluence, 9.5 to 11J/cm2 | Cheek, nose, and lips | Excellent improvement in all patients (75%-100%)Mean number of sessions to obtain response: 2.6 |
| Werner et al.,30 2008 | 4 | Nd:YAGPulse duration, 0.5-90msMaximum fluence, 450J/cm2Spot, 3-10mm | Case 1: cheeks, chin, and foreheadCase 2: fingertipsCase 3: fingers, ears, cheeks, chinCase 4: cheeks | Case 1: 50%-75% response at 8 weeksCase 2: 75%-95% response at 8weeks; adverse effects: white-greyish pigmentationCase 3: 75%-95% improvement in fingers after 2 sessions; 50% improvement in ears, chin, and cheeks with mild atrophy and hypopigmentationCase 4: 90% response after 8 weeks |
| Fernández-Jorge et al.,31 2007 | 3 | Photoderm-Vasculight system, which combined intense pulsed light + Nd:YAG 1064nm | Cheek, lips, nose | Complete response in all cases without any relapse during follow-up at 2 years in 2 of the 3 cases |
Argon lasers have been used for the treatment of cutaneous telangiectases; however, they have the disadvantage of causing scarring.32
Other studies have reported the use of intense pulsed light, followed by Nd:YAG laser pulses, with acceptable cosmetic outcomes.30
ConclusionsOsler-Weber-Rendu syndrome, or HHT, is a rare multisystemic disease with a broad clinical presentation and a substantial risk of complications, and so timely diagnosis and genetic counseling is required. Clinicians do not always have a high awareness of this condition and often do not recognize the disease until the onset of severe life-threatening complications.
Prognosis for survival of patients with HHT is favorable if complications are diagnosed and treated in time.
Laser treatment of cutaneous lesions has shown promise, although more long-term studies are required to more precisely determine their efficacy and to standardize the parameters according to the target site.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Orizaga-y-Quiroga TL, Villarreal-Martínez A, Jaramillo-Moreno G, Ocampo-Candiani J. Síndrome de Osler-Weber-Rendu y su relación con la dermatología. Actas Dermosifiliogr. 2019;110:527–532.


