



El diagnóstico de la neurofibromatosis 1 (NF1) plantea dificultades en niños sin antecedentes familiares durante la primera infancia. En este estudio pretendemos estimar la demora diagnóstica de los pacientes sin antecedentes familiares de NF1 y definir la repercusión de considerar las manchas café con leche y las efélides como un único criterio diagnóstico.
Pacientes y métodosEstudio observacional descriptivo retrospectivo en el que se revisaron los hitos diagnósticos de la NF1 en las historias clínicas de los pacientes menores de 18 años atendidos en nuestro centro. Distribuimos a los pacientes en dos grupos en función de la existencia de antecedentes de NF1 entre sus progenitores, considerando las manchas café con leche y las efélides como un único criterio y aceptando el estudio genético como criterio de confirmación en casos de elevada sospecha.
ResultadosSe incluyeron en el estudio 108 menores con diagnóstico de NF1. La edad media de diagnóstico en nuestra serie fue de 3,94 años (desviación estándar:±3,8 años). En el grupo 1, sin antecedentes, la edad media de diagnóstico fue de 4 años y 8 meses, mientras que en el grupo 2, con antecedentes, fue de 12 meses, siendo la demora en el diagnóstico de 3 años y 8 meses entre ambos grupos.
ConclusiónLas lesiones cutáneas representan, en la mayoría de los casos, las primeras manifestaciones clínicas de la enfermedad. Consideramos necesaria la actualización de los criterios diagnósticos del NIH con el fin de facilitar el diagnóstico en los primeros años de vida.
The neurofibromatosis 1 (NF1) diagnosis is challenging in young children without a family history of NF1. The aims of this study were to estimate diagnostic delays in children without a family history of NF1 and to examine the effects of using café au lait macules and skin fold freckling as a single diagnostic criterion.
Patients and methodsRetrospective, descriptive, observational study of all patients diagnosed with NF1 before the age of 18 years who were seen at our hospital. The medical records of those included were reviewed to identify the date on which the diagnostic criteria of NF1 were objectified. The patients were categorized into 2 groups: those with a known parental history of NF1 and those without. Café au lait macules and skin fold freckling were assessed as a single diagnostic criterion, and genetic evidence was considered to confirm highly suspicious cases.
ResultsWe studied 108 patients younger than the age of 18 years with a diagnosis of NF1. Mean (SD) age at diagnosis was 3.94 (±3.8) years for the overall group, 1 year for patients with a parental history of NF1, and 4 years and 8 months for those without. Diagnosis was therefore delayed by 3 years and 8 months in patients without a family history.
ConclusionSkin lesions were the first clinical manifestation of NF1 in most patients. We believe that the National Institutes of Health's diagnostic criteria for NF1 should be updated to aid diagnosis in young children.
El diagnóstico de la neurofibromatosis tipo 1 (NF1) (#162200) se basa en el reconocimiento de al menos 2 de los 7 criterios clínicos definidos por el National Institutes of Health (NIH) en 19871. Recientemente un grupo de expertos ha propuesto una modificación de estos criterios2 (tabla 1). Se estima que el diagnóstico de la NF1 proviene de los dermatólogos en el 50% de los casos3.
Criterios diagnósticos del NIH frente a los propuestos por Legius et al. en 2021
| Criterios diagnósticos de la NF1 del NIH | 1. Seis o más MCCL (mayores a 5mm en la infancia o mayores a 15mm tras la pubertad)2. Efélides axilares o inguinales. Signo de Crowe3. Dos o más neurofibromas de cualquier subtipo o un neurofibroma plexiforme4. Glioma de la vía óptica5. Dos o más nódulos de Lisch en la exploración oftalmológica6. Lesiones óseas características (displasia del esfenoides o pseudoartrosis de huesos largos)7. Antecedentes de un familiar de primer grado afecto |
| Revisión de los criterios propuestos por Legius et al. | A. Los criterios diagnósticos para la NF1 se cumplen en un individuo cuyos progenitores no tienen antecedentes de NF1 y que presentan dos o más de los siguientes criterios:1. Seis o más MCCL (diámetro mayor a 5mm en la infancia o mayores a 15mm tras la pubertad)2. Efélides axilares o inguinales. Signo de Crowe*3. Dos o más neurofibromas de cualquier subtipo o un neurofibroma plexiforme4. Glioma de la vía óptica5. Dos o más nódulos de Lisch en el iris en la exploración oftalmológica con lente de hendidura, o dos o más anormalidades coroideas definidas como nódulos parcheados brillantes observados en la tomografía de coherencia óptica de reflectancia próxima al infrarrojo6. Lesiones óseas características como la displasia esfenoides** el arqueamiento anterolateral de la tibia o la pseudartrosis de un hueso largo7. Identificación de una variante del gen NF1 patogénica heterocigota con una proporción del alelo mutado de más del 50% en tejido aparentemente normal como los glóbulos blancosB. El descendiente de un progenitor que cumple con los criterios diagnósticos especificados en el punto A, será diagnosticado de NF1, si presenta uno o más criterios incluidos en A |
MCCL: manchas café con leche; NIH: National Institutes of Health.
Aparecen en cursiva las incorporaciones de la revisión del grupo de expertos.
Si bien, el reconocimiento de estos criterios puede resultar sencillo en el adulto4, en niños pequeños sin antecedentes familiares, el diagnóstico de certeza suele demorarse varios años. La edad media de diagnóstico de la NF1 oscila entre los 2,65 y los 4,5 años en las series pediátricas de mayor tamaño muestral3,5–7. En ausencia de antecedentes familiares de NF1, la observación de múltiples manchas café con leche (MCCL) durante los primeros meses de vida, supone el primer criterio diagnóstico o criterio de sospecha de la enfermedad en la práctica totalidad de los casos.
Con este estudio pretendemos estimar la demora diagnóstica de los pacientes sin antecedentes familiares de NF1 y estimar la repercusión de la consideración de las MCCL y las efélides como un único criterio diagnóstico (criterio pigmentario).
Material y métodosEstudio observacional descriptivo retrospectivo en el que se revisaron las historias clínicas de todos los pacientes menores de 18 años con diagnóstico de NF1 atendidos entre el 1 mayo de 2012 y el 30 de abril de 2016 en nuestra unidad de NF1 pediátrica. Los criterios de inclusión requerían que verificaran dos o más criterios del NIH o 2 criterios más la detección de mutaciones en el gen NF1 para aquellos que presentaran solamente MCCL y efélides y la obtención del consentimiento informado por parte de los tutores del paciente.
El protocolo del estudio fue revisado y aprobado por el comité de ética e investigación del Hospital Niño Jesús de Madrid, R-0024/16 Acta n.° 11/16, 27 de septiembre de 2016.
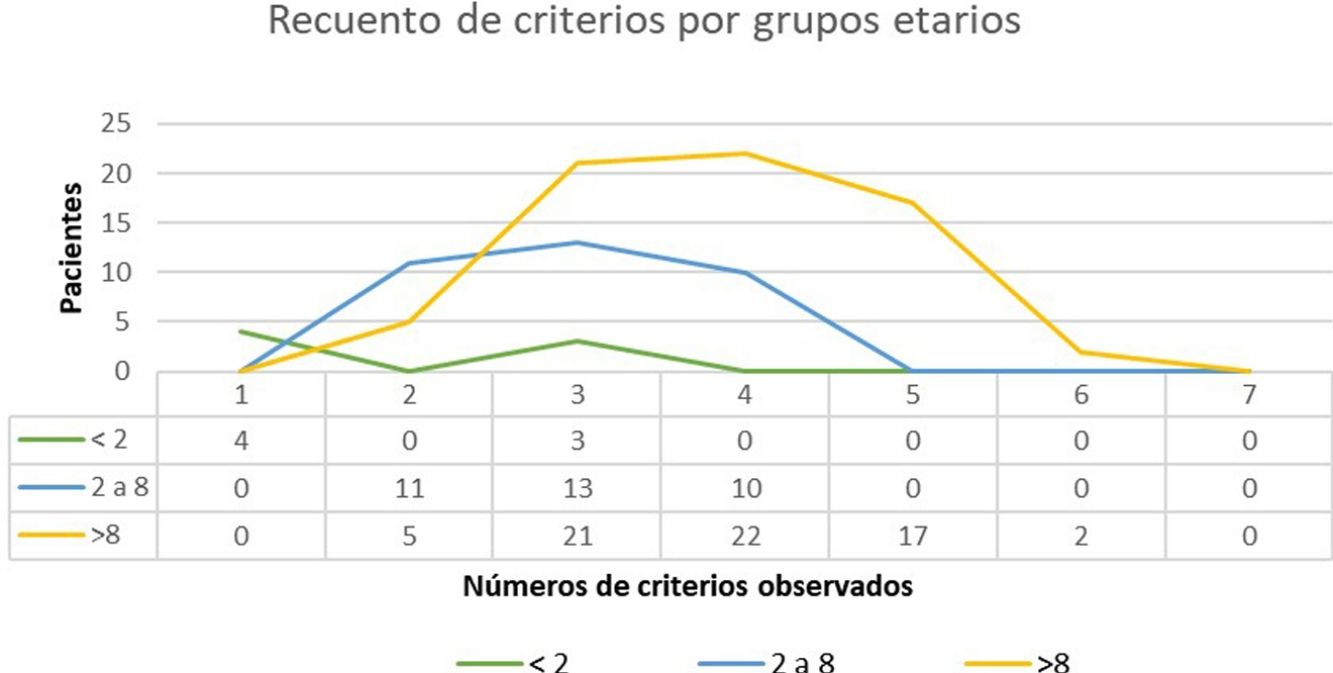
ResultadosSe atendieron un total de 135 niños con sospecha de NF1, de los cuales se incluyeron en el estudio 108. La edad media de diagnóstico en nuestra muestra fue de 3,94 años (desviación estándar [DE] de±3,8 años) y la mediana 3 años. Clasificamos en 2 grupos a los pacientes en función de la existencia o no de antecedentes familiares de NF1. En el grupo 1 se incluyeron 86 pacientes (79,63%), sin antecedentes familiares de NF1 conocidos o con mutación «de novo». En este grupo, el criterio de 6 o más MCCL>5mm supuso el criterio de sospecha o primer criterio diagnóstico registrado en todos los casos. La edad media de este grupo fue de 4,7 años (4 años y 8 meses, DS±3,87) y una edad mediana de 4 años. En 3 de los casos incluidos en el grupo 1, se alcanzó el diagnóstico definitivo al descubrir la enfermedad en uno de los progenitores del paciente el día de la consulta, diagnóstico que se desconocía hasta ese momento. El estudio genético confirmó la sospecha diagnóstica en 19 casos (17,59%) que contaban exclusivamente con criterios pigmentarios (tabla 2). En el grupo 2 (22, 20,37%) incluimos a los pacientes con algún progenitor diagnosticado de NF1. El promedio de edad de diagnóstico en estos casos fue de 0,99 años (12 meses, DE:±1,31) y la mediana de 0,62 años (7,5 meses). El resto de las características de la población se resumen en la tabla 2. En la figura 1 se recoge el número de criterios que observamos en los pacientes en función del grupo de edad. Entre los menores de 2 años incluimos aquellos que además de criterios pigmentarios aportaban un estudio molecular que demostraba una variante patogénica del gen NF1. En la mayoría de los pacientes se identificaron 3 o 4 criterios y en ninguno se registraron los 7 criterios (fig. 1).
Distribución de criterios diagnósticos del NIH. Edades media y mediana del diagnóstico
| Criterio sospecha | Criterio confirmación | n - % | x¯ | Mediana | DE | |
|---|---|---|---|---|---|---|
| Grupo 1n=86, (79,63%) | MCCL±efélides | Antecedentes familiares sin diagnóstico previo | 3, 2,79% | 7,08 | 10 | 5,94 |
| x¯: 4 años y 8 meses, DE±3,87 | Neurofibromas | 23, 21,39% | 5,71 | 4 | 5,16 | |
| GVO | 16, 14,81% | 3,38 | 3 | 1,68 | ||
| Nódulos de Lisch | 24, 22,32% | 5,94 | 6 | 3,51 | ||
| Displasia ósea | 1, 0,93% | 1,5 | 1,5 | NV | ||
| «Estudio genético» | 19, 17,59% | 2,96 | 2,5 | 2,73 | ||
| Grupo 2 | Antecedentes familiares de 1.er grado | MCCL | 19, 17,59% | 0,88 | 0,5 | 1,28 |
| n=22, (20,37%) | Efélides | 1, 0,93% | 0,5 | 0,5 | NV | |
| x¯:12 meses, DE±1,31 | Neurofibroma | 1, 0,93% | 3,5 | 3,5 | NV | |
| «Estudio genético» | 1, 0,93% | 1 | 1 | NV |
DE: desviación estándar; GVO: glioma de la vía óptica; MCCL: manchas café con leche; NV: no valorable.

Recuento de criterios verificados por grupos etarios. En el grupo etario de los menores de 2 años incluidos en el estudio solamente 4 fueron diagnosticados presentando MCCL y antecedentes familiares y 3 niños que presentaban 3 criterios. En la mayoría de los pacientes en la edad pediátrica se identifican 3 o 4 criterios y en ninguno se observaron los 7 criterios.
Durante el periodo del estudio pudimos observar NA en 22 pacientes y XGJ en 3 de ellos con anterioridad al establecimiento del diagnóstico definitivo. En este subgrupo la edad media de observación de los NA fue de 3,1 años [3 meses-14 años] (±3,6). Además, cabría resaltar que 14 de estos pacientes presentaron NA antes de presentar efélides en los grandes pliegues (tabla 3).
Edad y orden de aparición de los criterios diagnósticos en los pacientes con NA o XGJ observados antes de alcanzar el diagnóstico definitivo de NF1
| Paciente | Sexo | Orden y edad de observación de los criterios diagnósticos y de los NA (años o meses) | Edad del diagnóstico definitivo | ||||
|---|---|---|---|---|---|---|---|
| 1.o | 2.o | 3.o | 4.o | 5.o | |||
| 1 | F | MCCL (18 m) | Efélides (2) | NA (2) | Nódulos Lisch (7) | Mutación NF1 (7) | 7 |
| 2 | M | MCCL (5 m) | NA (18 m) | Efélides (2) | Mutación NF1 (3) | - | 3 |
| 3 | M | MCCL (6 m) | NA (6 m) | NFP (ECO) (1) | Mutación NF1 (1)- | - | 1 |
| 4 | M | MCCL (6 m) | Efélides (6 m) | NA (9) | NF x2 (9) | Mutación NF1 (9) | 9 |
| 5 | F | MCCL (7 m) | NA (7 m) | Mutación NF1 (1) | - | - | 1 |
| 6 | F | MCCL (9 m) | NA (9 m) | Nódulos Lisch (2) | Mutación NF1 (3) | Efélides (3) | 2 |
| 7 | M | MCCL (6 m) | Efélides (8m) | NA (9 m) | Mutación NF1 (1) | - | 1 |
| 8 | M | MCCL (4) | NA (4) | Mutación NF1 (4) | Efélides (4) | - | 4 |
| 9 | M | MCCL (3 m) | NA (3 m) | Mutación NF1 (1) | Efélides (7) | NFP- | 1 |
| 10 | F | MCCL (4 m) | Efélides (1) | NA (3) | NFP (ECO) (4) | Mutación NF1 (4) | 4 |
| 11 | M | MCCL (9 m) | Efélides (NC) | NA (5) | Mutación NF1 (7) | - | 7 |
| 12 | M | MCCL (6 m) | NA/XGJ (9/18 m) | Mutación NF1 (12m) | Efélides (18m) | GVO (2) | 1 |
| 13 | F | MCCL (2) | NA (9) | Efélides (10) | Mutación NF1 (11) | - | 11 |
| 14 | F | MCCL (6 m) | Efélides (NC) | NA (5) | Mutación NF1 (7) | NFx2 (ECO) (8) | 7 |
| 15 | F | MCCL (12 m) | NA (12 m) | Mutación NF1 (2) | NF x 2 (4) | - | 2 |
| 16 | M | MCCL (6 m) | NA (6 m) | Mutación NF1 (1) | - | - | 1 |
| 17 | F | MCCL (9 m) | NA /XGJ (9/11 m) | Mutación NF1 (1) | - | - | 1 |
| 18 | F | MCCL (3 m) | XGJ/NA (18 m) | Mutación NF1 (6) | Nódulos Lisch (6) | NF x 2 (6) | 6 |
| 19 | M | MCCL (1) | NA (1) | Mutación NF1 (4) | Efélides (5) | - | 4 |
| 20 | M | MCCL (1) | Efélides (5) | NA (5) | NFx2 (ECO) (6) | - | 6 |
| 21 | M | MCCL (NC) | Efélides (NC) | NA (14) | NFx2 (ECO) (15) | - | 15 |
| 22 | F | MCCL (2 m) | NA (3 m) | Nódulos Lisch (12) | - | - | 12 |
ECO: ecografía; GVO: glioma de la vía óptica; MCCL: manchas café con leche; NA: nevus anémicos; NC: no conocida; NF: neurofibroma; NFP: neurofibroma plexiforme; XGJ: xantogranuloma juvenil.
El criterio de mayor transcendencia en el diagnóstico de la NF1 y el principal motivo de sospecha de la enfermedad, desde los primeros meses de vida, son las MCCL. En pacientes de edad avanzada las MCCL pueden aclararse y desaparecer8 y en las formas de NF1 espinal familiar también pueden estar ausentes9. En nuestra serie en todos los pacientes se observaron al menos 6 de estas manchas, incluyendo los pacientes menores de 2 años. Se estima que entre el 66 y el 99% de los pacientes afectos por NF1 presentan 6 o más MCCL en el primer año de vida10,11. La identificación de múltiples MCCL en un recién nacido supone un importante motivo de derivación a dermatología por parte de los pediatras de atención primaria. A partir de ese momento, en ausencia de antecedentes familiares, se inicia la espera para la confirmación del diagnóstico11. Se estima que esta demora dura entre 2 y 3 años12 y que solamente entre el 20 y el 46% de los niños sin antecedentes familiares de NF1 son diagnosticados antes de los 2 años de edad3,7. En nuestra serie solo el 6,48% de los pacientes cumplían los criterios antes de cumplir 2 años. Algunos autores consideran que la constatación de las MCCL «típicas», de morfología redondeada u oval, color marrón homogéneo y bordes regulares bien definidos y mayores a 5mm en los primeros meses de vida, es un hallazgo con un elevado valor predictivo positivo13. En ausencia de otros hallazgos, la mayoría de los facultativos se decantan por el seguimiento clínico del paciente o por solicitar un estudio molecular, que tampoco está exento de demoras y costes económicos. La diferencia en la edad media de diagnóstico entre los pacientes con antecedentes familiares y los esporádicos en nuestra serie fue de 3 años y 8 meses. Esta espera puede resultar angustiosa para los progenitores y puede retrasar la detección de complicaciones3.
Las efélides también están presentes en la amplia mayoría de los pacientes pediátricos en la NF1, en concreto entre el 85,3 y el 93,7% las presentan5,6, aunque otros estudios retrospectivos reducen dicha frecuencia al 21,1%7. Observamos estas efélides en un 87,5% de nuestros pacientes, pese a lo cual en solo uno de los casos se presentaron con anterioridad al cumplimiento del criterio de MCCL y tuvieron influencia real en el diagnóstico. Numerosos autores entienden que las efélides son MCCL de menor tamaño, histopatológicamente indiferenciables de las MCCL y por lo tanto no deberían considerarse un criterio independiente. La descripción de otras genodermatosis que también se manifiestan con MCCL y efélides como el síndrome de Legius, el síndrome de constitutional mismatch repair deficiency, o el síndrome de Noonan con múltiples lentigos (LEOPARD) condiciona el diagnóstico basado solamente en los criterios pigmentarios14.
En nuestra muestra, la edad media de diagnóstico (3,94 años), se incluye en el rango de edades medias de diagnóstico reflejado en la literatura (2,65-4,5 años)3,5,6, pese a requerir la observación de un tercer criterio o la detección de la mutación patológica causal en el estudio genético para la inclusión de los pacientes que solamente presentaban MCCL y efélides. Aunque la posibilidad de incurrir en falsos positivos en pacientes con criterios pigmentarios cutáneos es muy baja4, entendemos que el signo de Crowe apoya, pero no confirma el diagnóstico de la NF115.
En la NF1, la mayoría de los criterios no son verificados hasta la etapa escolar11. En el caso de las efélides, los neurofibromas y los nódulos de Lisch el porcentaje de cumplimiento de los criterios se incrementa con el paso de los años. Los gliomas de la vía óptica (GVO) y las señales hiperintensas en la resonancia nuclear magnética son más frecuentes en el estrato de 2 a 8 años para disminuir en el de mayores de 8 años. Paradójicamente, hallazgos específicos y con elevado valor predictivo positivo que no son considerados como criterio por algunos expertos2, se manifiestan de forma temprana, y por lo tanto, serían de mayor utilidad diagnóstica. Este es el caso de los xantogranulomas juveniles (XGJ) y de los nevus anémicos (NA) cuya prevalencia es mayor en el grupo de menores de 2 años14. Nuestros resultados previamente publicados demuestran que la totalidad de los niños afectos por los criterios pigmentarios y NA (22) o XGJ (3), fueron posteriormente diagnosticados de NF114 y la edad media de observación de los NA en este subgrupo fue considerablemente menor a la edad media de diagnóstico (3,1 frente a 3,94 años) e incluso inferior a la de las efélides axilares.
El tercer criterio dermatológico más frecuente está representado por los neurofibromas. Los neurofibromas son tumores benignos que contienen todos los elementos celulares del nervio periférico. Se estima en estudios retrospectivos que en la edad pediátrica entre el 38,1 y el 38,4% presentan al menos dos lesiones cutáneas o subcutáneas y entre el 23 y el 24,7% un neurofibroma plexiforme (NFP)5,7. Estos datos son ligeramente superiores a los obtenidos en nuestro estudio, donde registramos un 37,19% y un 20,37% respectivamente. Un 20,37% adicional presentó una única lesión compatible con un neurofibroma, que no pudo cuantificarse como criterio al ser lesiones aisladas o por no haber confirmado el patrón plexiforme.
Los neurofibromas en los niños se caracterizan por una marcada variabilidad clínica, motivo por el cual, decidimos caracterizarlos y clasificarlos basándonos en nueve patrones clínico-ecográficos con un elevado índice de correlación interobservador a partir de un análisis de clústeres16. Los más fáciles de reconocer son los denominados neurofibromas cutáneos «clásicos» que suelen manifestarse como pequeñas pápulas o nódulos del color de la piel normal o ligeramente hiperpigmentados, de tacto blando, que durante su evolución adquieren un aspecto sésil o pediculado17. Suelen detectarse de forma nítida a partir de los 6 años11, aunque pueden observarse como pequeñas pápulas o tenues elevaciones circunscritas sin alteración de la pigmentación a edades más tempranas, hecho que nos permitiría confirmar el diagnóstico anticipadamente empleando la ecografía cutánea o tomando una biopsia en casos dudosos16.
Aunque son escasas las referencias a las manifestaciones de la NF1 durante el periodo perinatal18, la identificación temprana de un NFP nos permitirá establecer el diagnóstico de la NF1 incluso desde los primeros meses de vida19. Los NFP se definen como lesiones congénitas que constituyen una variante clínicopatológica en sí misma y se les considera una lesión patognomónica de la NF120–22. La presencia de grandes MCCL de bordes irregulares de aparición congénita o durante los primeros meses de vida es sugestivo de NFP y nos brinda la posibilidad de diagnosticar precozmente a los pacientes si conseguimos establecer un diagnóstico diferencial mediante biopsias o ecografía entre las mal llamadas MCCL atípicas y los nevus melanocíticos congénitos16,21. Al igual que Peltonen et al., consideramos que se debería requerir la constatación histopatológica o ecográfica del tipo de neurofibroma23.
Los dermatólogos podemos identificar otros signos clínicos considerados criterios. En concreto los nódulos de Lisch suponen un elemento clave en el diagnóstico de la NF1. Supusieron el criterio de confirmación en el 22% de nuestros casos. Estos hamartomas iridianos suelen observarse a partir de los 5 años y su identificación con lámpara de hendidura está condicionada a la experiencia del oftalmólogo pediátrico y a la colaboración del paciente, con el consiguiente detrimento de la sensibilidad y especificidad en niños pequeños5. Los nódulos de Lisch también se visualizan con facilidad en pacientes de ojos claros mediante dermatoscopia.
La frecuencia estimada del glioma de la vía óptica GVO es de aproximadamente del 5-15%24, en nuestra serie la incidencia de GVO ascendió al 25,93%. Los GVO supusieron la confirmación del diagnóstico en el 14,81% de los pacientes. Consideramos que la alta prevalencia de GVO observada en nuestra serie se justifica por el protocolo de los neurólogos pediátricos de nuestra unidad que contempla el empleo de la resonancia nuclear magnética en pacientes asintomáticos con criterios pigmentarios de NF1 a partir de los 2 años.
Las alteraciones óseas son los hallazgos de menor prevalencia. Pese a que pueden manifestarse a cualquier edad, el porcentaje de casos en nuestra serie no difiere de los registrados en la literatura25. Podemos sospechar la existencia de anomalías óseas si existen alteraciones de la marcha o al observar curvaturas de los miembros inferiores.
Las nuevas técnicas de diagnóstico genético molecular, como el Next Generation Sequencing, alcanzan una precisión diagnóstica del 95%26. Por lo tanto, su inclusión como criterio no suscita ninguna controversia. En la mayoría de los casos la detección de una mutación concreta carece de valor pronóstico, aunque puede establecerse una relación genotipo-fenotipo que facilite el seguimiento de algunos pacientes27.
Menos del 25% de nuestros pacientes presentaban antecedentes familiares de la enfermedad, a pesar de que por consenso se considera que la mitad de los casos son hereditarios7. Es probable que los progenitores conocedores de las manifestaciones de la enfermedad no consultan en dermatología o no sean derivados en ausencia de comorbilidades. En última instancia, pero no menos importante, consideramos que la menor incidencia de casos familiares pueda deberse al fruto del consejo genético y al empleo de las técnicas de diagnóstico genético preimplantacional28.
Las principales limitaciones de este estudio son su diseño observacional retrospectivo y el reducido tamaño muestral de niños menores de 2 años. Destacar también que se hace referencia exclusivamente a la edad en la que se objetivaron las distintas manifestaciones de la enfermedad por parte de un especialista, pudiendo coincidir o no con la edad de presentación o la referida por los progenitores.
Coincidimos con Legius et al.2 en la necesidad de la actualización de los criterios diagnósticos del NIH. Sin embargo, consideramos que en su propuesta, el único cambio enfocado a reducir la demora diagnóstica de la NF1, es la inclusión de la detección de variantes patogénicas en el gen NF1 mediante el análisis molecular29. Cabría destacar que el estudio genético ya era considerado un criterio diagnóstico de facto en la práctica clínica diaria5,30, y a tenor de nuestros resultados, indicar el estudio molecular en casos dudosos no supone un gran impacto en la edad media de diagnóstico7,14. Por lo tanto, consideramos oportuno evaluar de nuevo la inclusión como criterios diagnósticos de los NA y XGJ, al ser manifestaciones infrecuentes en otras rasopatías, que se suelen presentar en la NF1 antes de los dos años de vida. En consecuencia, consideramos de especial relevancia que en los equipos multidisciplinares dedicados a la atención de pacientes con neurofibromatosis se potencie la participación de los dermatólogos.
FinanciaciónNo se recibió ninguna financiación para la realización de este trabajo. La tesis que recoge parte de la información contenida en este trabajo fue premiada con el Accésit a la Mejor Tesis publicada en 2017 de la Sección Centro de la AEDV.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.
Este estudio se incluye en la tesis doctoral titulada: «Estudio clínico ecográfico de la neurofibromatosis tipo 1 en la edad pediátrica».



