



El carcinoma sebáceo (CS) es una neoplasia cutánea maligna poco común pero potencialmente agresiva, que a menudo se clasifica por su ubicación anatómica en ocular y extraocular. Ambos tumores se comportan de manera diferente y pueden manifestarse como neoplasias primarias o secundarias asociadas al síndrome de Muir Torre (SMT)1.
Se revisaron los pacientes diagnosticados de CS registrados en la base de datos del servicio de Anatomía Patológica del Hospital Universitario Fundación Alcorcón desde enero de 1998 a diciembre del 2022. Los datos fueron obtenidos a partir de la revisión de todas las historias clínicas y biopsias, analizando sus características demográficas, clínicas, histológicas, tratamiento y evolución.
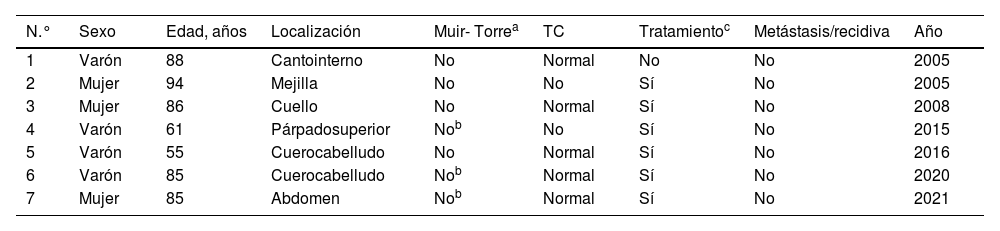
Se identificaron 7 casos con CS diagnosticados en los últimos 24 años. Las características más relevantes se sintetizan en la tabla 1. La edad mediana de los pacientes fue de 85 años (rango entre 55 y 94), siendo 4 de ellos varones y 3 mujeres. En 6de los casos, el tumor se localizaba en cabeza y cuello, y 2eran perioculares. Clínicamente, se presentaron como pápulas o nódulos eritemato-anaranjados, con una mediana de 2cm (rango entre 0,4 y 5cm) (fig. 1A-C). La mediana hasta el diagnóstico fue de 5 meses (rango entre 3 y 36 meses). Para el diagnóstico fue necesaria la confirmación histológica de células neoplásicas con diferenciación sebácea, con grados variables de diferenciación y atipia, apoyándose de tinciones inmunohistoquímicas como la adipofilina, receptores de andrógenos o citoqueratina 5/6 (fig. 1D-F).
Características demográficas más relevantes
| N.° | Sexo | Edad, años | Localización | Muir- Torrea | TC | Tratamientoc | Metástasis/recidiva | Año |
|---|---|---|---|---|---|---|---|---|
| 1 | Varón | 88 | Cantointerno | No | Normal | No | No | 2005 |
| 2 | Mujer | 94 | Mejilla | No | No | Sí | No | 2005 |
| 3 | Mujer | 86 | Cuello | No | Normal | Sí | No | 2008 |
| 4 | Varón | 61 | Párpadosuperior | Nob | No | Sí | No | 2015 |
| 5 | Varón | 55 | Cuerocabelludo | No | Normal | Sí | No | 2016 |
| 6 | Varón | 85 | Cuerocabelludo | Nob | Normal | Sí | No | 2020 |
| 7 | Mujer | 85 | Abdomen | Nob | Normal | Sí | No | 2021 |
TC: toraco-abdomino-pélvico.
 Imagen clínica de 3 pacientes. A) Lesión proliferativa, exofítica, eritematosa, friable y ulcerada de 5cm en hemiabdomen izquierdo. B) Lesión exofítica, pediculada, eritematosa, friable, y erosionada, de 4×2cm localizada en la cara lateral derecha cervical. C) Tumoración eritematosa, ulcerada, friable, de 2cm localizado en mejilla derecha. D-F) Imagen histopatológica. Hematoxilina eosina. Panorámica. Proliferación neoplásica exofítica con componente in situ asociado en el margen izquierdo (D). Hematoxilina eosina ×400. Áreas de carcinoma sebáceo in situ compuestas por sebocitos maduros e inmaduros (E). Tinción de inmunohistoquímica con adipofilina ×40. Inmunoexpresión para adipofilina en los sebocitos maduros (F).")
A-C) Imagen clínica de 3 pacientes. A) Lesión proliferativa, exofítica, eritematosa, friable y ulcerada de 5cm en hemiabdomen izquierdo. B) Lesión exofítica, pediculada, eritematosa, friable, y erosionada, de 4×2cm localizada en la cara lateral derecha cervical. C) Tumoración eritematosa, ulcerada, friable, de 2cm localizado en mejilla derecha. D-F) Imagen histopatológica. Hematoxilina eosina. Panorámica. Proliferación neoplásica exofítica con componente in situ asociado en el margen izquierdo (D). Hematoxilina eosina ×400. Áreas de carcinoma sebáceo in situ compuestas por sebocitos maduros e inmaduros (E). Tinción de inmunohistoquímica con adipofilina ×40. Inmunoexpresión para adipofilina en los sebocitos maduros (F).
Dos pacientes presentaban antecedentes personales oncológicos, sin antecedentes familiares, un carcinoma renal nefrectomizado, y un diagnóstico simultáneo de melanoma, carcinoma epidermoide y CS.
Se realizó una TC toraco-abdomino-pélvico como estudio de extensión en 5 de los 7 pacientes, sin encontrar hallazgos patológicos. Con la excepción de un paciente fallecido antes de la cirugía por sepsis, el tratamiento fue quirúrgico, realizándose una extirpación amplia hasta plano fascial con márgenes clínicos de 1cm. La mediana de seguimiento de los pacientes fue de 2 años (rango entre 6 meses y 14 años), no identificándose hasta la fecha recidivas ni metástasis en ninguno de los pacientes. Dos pacientes fallecieron durante el primer año tras el diagnóstico por motivos infecciosos (neumonía adquirida). Ninguno de los pacientes cumplió criterios de SMT según los principios de Mayo2 (tabla 2). Se realizó estudio de inestabilidad de microsatélites en el 3 de los casos, siendo todos ellos negativos.
Puntuación de riesgo de mayo del síndrome Muir-Torre
| Variable | Puntuación |
|---|---|
| Edad en el momento del diagnóstico (años)a | |
| 60 años o más 0 | 0 |
| Menor de 60 años 1 | 1 |
| Número total de neoplasias sebáceas | |
| 1 | 0 |
| 2 o más | 2 |
| Antecedentes personales de algún cáncer relacionado con el síndrome de Lynchb | |
| No | 0 |
| Sí | 1 |
| Antecedentes familiares de cualquier cáncer relacionado con el síndrome de Lynch | |
| No | 0 |
| Sí | 1 |
Puntuación de riesgo de mayo del síndrome Muir-Torre. Las puntuaciones de las 4variables se suman para crear una puntuación total, con un rango posible de 0 a 5. Una puntuación de 2 o más tiene una sensibilidad del 100% y una especificidad del 81% para predecir una mutación en la línea germinal de un síndrome de Muir-Torre.
El CS tiene una tasa de incidencia estimada de 2,4 casos por millón de individuos por año3. Se localiza preferentemente en la región periocular, afectando hasta en el 75% de los casos a esta localización4. Es ligeramente más frecuente en varones (58%), con una media de edad de 67,9 años1.
Suele presentarse como un nódulo o pápula ulcerada, indolora, de color rojo-anaranjado, que puede simular otras entidades (blefaritis, chalazión, etc.), por lo que el diagnóstico definitivo suele retrasarse, aumentando la morbimortalidad1,4-6.
El diagnóstico requiere la confirmación histológica de la diferenciación sebácea. Los tumores bien diferenciados muestran células claras microvacuoladas con núcleos grandes, vesiculosos o hipercromáticos con nucléolos prominentes, siendo escasas o raras en los moderada o pobremente diferenciados. Debido a la similitud con otras neoplasias cutáneas la inmunohistoquímica puede ser de gran ayuda (EMA [tumores bien diferenciados], receptores de andrógenos [tinción nuclear], adipofilina [vacuolas citoplasmáticas] y citoqueratinas de alto peso [CK 5/6])1,4-6.
La escisión quirúrgica se acepta como el pilar del tratamiento del CS tanto ocular como extraocular, siendo la cirugía micrográfica de Mohs el tratamiento de primera línea1,3-6. La tasa de supervivencia global a 5 años del carcinoma sebáceo es del 78% para la enfermedad localizada y regional y del 50% para la enfermedad metastásica, por lo que está indicado un seguimiento estrecho de los pacientes, recomendándose revisiones cada 6 meses durante 3 años y posteriormente anuales1,3.
El SMT es una variante fenotípica del síndrome de cáncer colorrectal hereditario sin poliposis (síndrome de Lynch) causada por mutaciones heredadas de forma dominante en los genes de reparación de errores del ADN y definida por una mayor predisposición al desarrollo de neoplasias sebáceas7-9. Aunque recomendaciones previas aconsejaban el estudio de inestabilidad de microsatélites (MSH1, MSH2, MLH1, PMS2) en todos los pacientes con CS3, en las últimas guías no está indicada la realización de cribado de SMT en CS perioculares, recomendándose individualizar en los extraoculares. Las publicaciones más recientes recomiendan un cribado inicial con criterios clínicos mediante la puntuación de riesgo de Mayo2,10.
En conclusión, la baja incidencia del CS y su clínica inespecífica nos debe alertar sobre la necesidad de establecer una alta sospecha clínica para evitar retrasos diagnósticos y terapéuticos, siendo de vital importancia el estudio histológico. Cabe destacar que, aunque el screening rutinario de SMT no está recomendado, la inmunohistoquímica es una prueba complementaria económica y disponible en la mayoría de los laboratorios de España, debiéndose plantear en todo paciente con carcinoma sebáceo. Nosotros, de acuerdo con las últimas guías, sugerimos un screening inicial con la puntuación de riesgo de Mayo, inmunohistoquímica y posteriormente estudio genético si se cumplen criterios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


