




Dystrophic epidermolysis bullosa pruriginosa (DEB-Pr) is a rare subtype of epidermolysis bullosa (EB) due to mutations to the COL7A1 gene that codifies collagen VII that causes dysfunctional anchoring fibers of the dermoepidermal junction. Histopathologically, subepidermal blisters are a common finding, along with clinically intensely pruritic, often pretibial lichenified plaques.
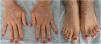
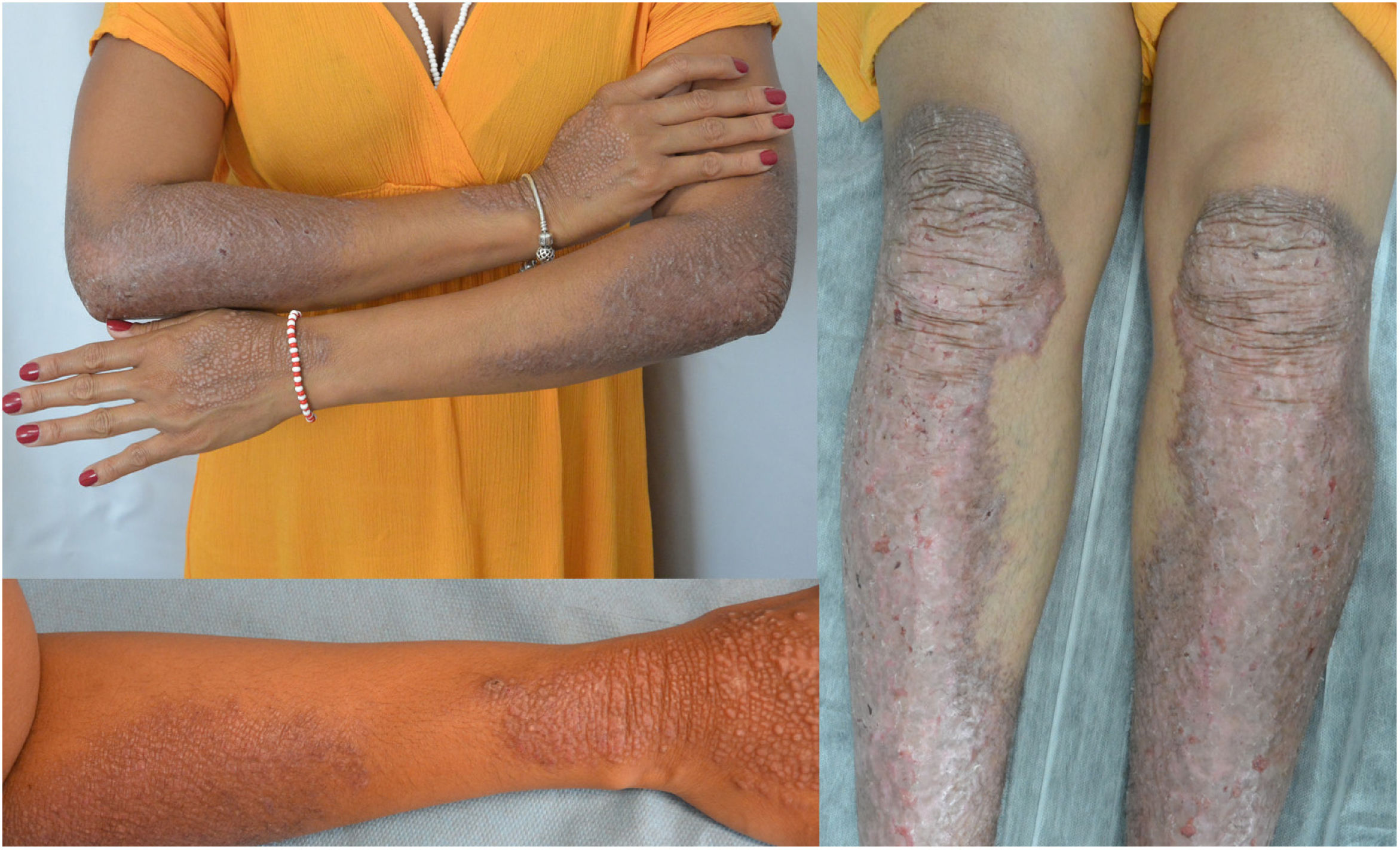
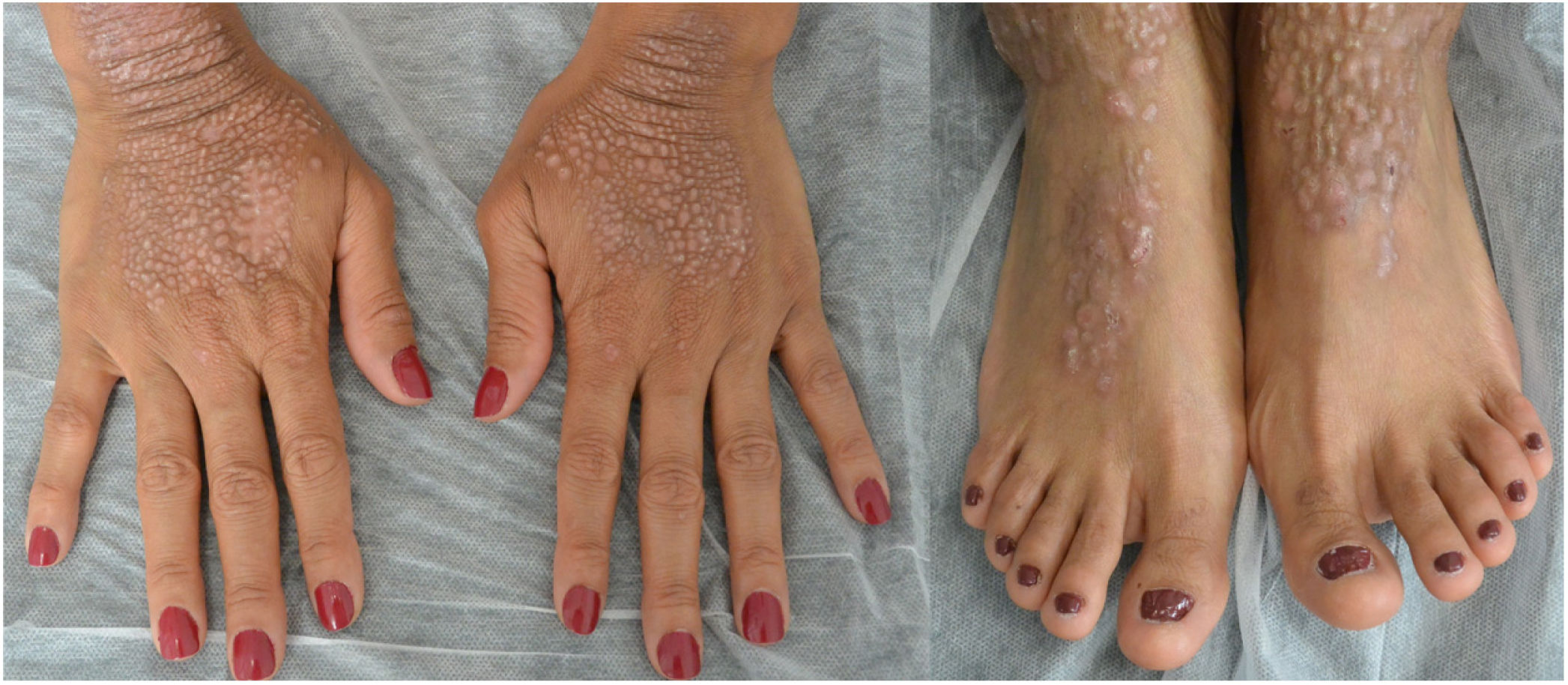
This is the case of a 34-year-old woman without a significant past medical history with intensely pruritic lesions on the back of her hands, forearms, and anterior side of both her legs since the age of 3. She occasionally noticed vesicles and blisters, especially after sustaining traumas. Her maternal stepbrother showed similar lesions, yet her parents (who were not blood relatives), or daughter were unaffected. The physical examination revealed the presence of well-demarcated brownish papules and lichenified plaques with multiple erosions of the distal extremities, along with toenail dystrophy (Figures 1 and 2).
The histopathological examination of skin biopsy revealed the presence of subepidermal blisters with minimal inflammation, and a negative direct immunofluorescence testing for immunoglobulins and complement. Electron microscopy revealed the presence of cleavage lines underneath the dense layer with loss of skin anchoring fibrils. The lab test results showed elevated levels of IgE (800 IU/mL). Following this plethora of findings, the patient was diagnosed with DEB-Pr.
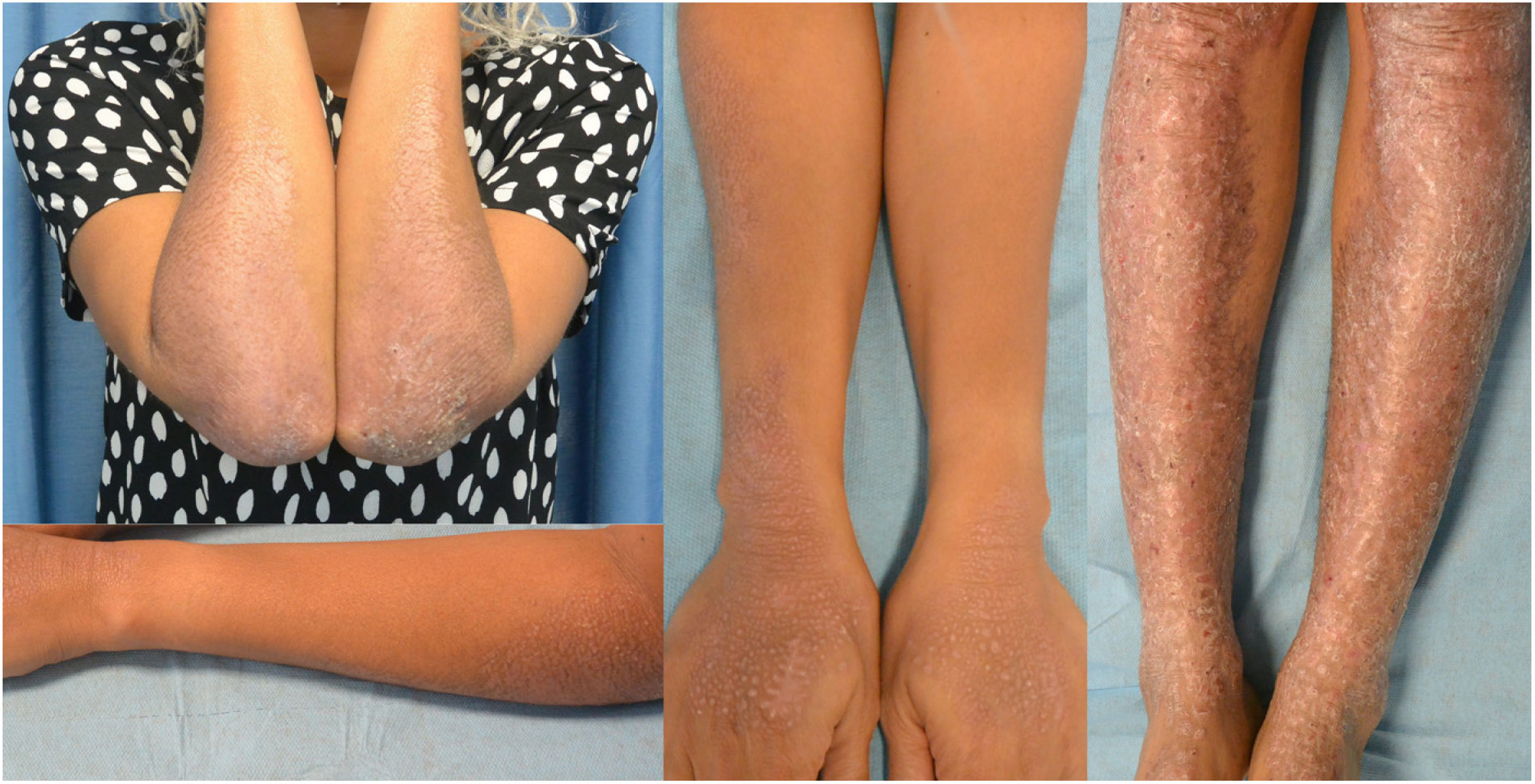
The patient had previously been treated with multiple topical (corticosteroids, calcineurin inhibitors, vitamin D analogs, doxepin, and compounded topical amitriptyline), and systemic drugs (thalidomide, cyclosporine, gabapentin, mirtazapine, and various oral antihistamines) without itching or skin lesion improvement. Since this clinical presentation had a significant impact on the patient's quality of life (DLQI score of 13), she was put on dupilumab on a compassionate use basis at an initial dose of 600mg followed by 300mg every 2 weeks. After 14 days, the patient reported significant relief from the itching, and 2 months into treatment, skin lesions improved. Nine months into treatment, the patient's quality of life had improved significantly (DLQI score of 5) and her skin lesions—especially on her upper limbs—had improved significantly too (figure 3). The only side effect noted was mild leukopenia, which resolved spontaneously without down-titration.
DEB-Pr was first reported by McGrath et al. back in 1994.1 Although DEB-Pr is a rare disease of autosomal dominant inheritance pattern, sporadic and recessive cases have been reported. The most common mutation of COL7A1 gene involves the substitution of glycine. However, to date, a clear genotype-phenotype correlation has not been established yet.2 Because of the wide clinical variability, even within the same family, factors such as atopy, iron deficiency, or elevated IgE levels have been hypothesized as modifiers. Also, 3 cases have been described associated with scabies.3
The onset of DEB-Pr can vary, with cases reported even in individuals older than 70 years.4 However, the onset of DEB-Pr often starts in adolescence with intense, uncontrollable itching, primarily affecting the distal extremities, and subsequently triggering papules, nodules, and lichenified plaques, which may exhibit a linear distribution, excoriations, and scarring. Although truncal distribution can occur, the face and flexural areas are often spared, while the most intense itching is found in the pretibial region. The appearance of blisters after trauma and the finding of toenail dystrophy are common findings. A total of 3 cases of asymptomatic cases have been reported to this date.5
Histopathologically, DEB-Pr is characterized by hyperkeratosis with acanthosis and subepidermal blisters located above the basement membrane, along with an interstitial and perivascular lymphocytic infiltrate. Direct immunofluorescence often tests positive for collagen VII antibodies, with a linear staining pattern along the basement membrane. As it happens with other EBs, electron microscopy shows changes to the dermoepidermal junction and fewer anchoring fibers. While genetic testing would confirm the diagnosis, DEB-Pr can be diagnosed based on typical clinical signs, the presence of a family history, compatible histopathology, and electron microscopy findings.6
Diagnosis can require a high level of suspicion, because some patients may develop the condition during adulthood with few lesions, being the presence of intact blisters a rare finding.7 Mainly, differential diagnosis should include prurigo nodularis, lichen simplex chronicus, and hypertrophic lichen planus, being histopathology examination a helpful tool.
Various treatments have been used to treat DEB-Pr, including topical, intralesional, and oral corticosteroids, topical calcineurin inhibitors, cryotherapy, UVB phototherapy, oral antihistamines and retinoids, dapsone, gabapentin, thalidomide, cyclosporine, mizoribine, tofacitinib, baricitinib, IV immunoglobulins, naltrexone, mirtazapine, and sertraline with little success, or only a few isolated successful cases. Although dupilumab—a full human monoclonal antibody that inhibits the alpha subunit of the interleukin-4 receptor—has been approved to treat atopic dermatitis, it can treat prurigo nodularis successfully, a condition that shares clinical characteristics with DEB-Pr. A total of 6 cases of patients treated with dupilumab have been described so far. In all of them itching and skin lesions improved without any side effects being reported. Up-titration was needed only in 1 patient (300mg/week).8
This was the case of a patient with DEB-Pr refractory to multiple treatments who improved her quality of life and skin lesions with the use of dupilumab. DEB-Pr should be suspected in patients with pruritic lesions predominantly affecting the legs.
Conflicts of interestNone declared.
We wish to thank Dr. Daniel Cameselle and Dr. Rafael Camacho Galán, as well as to the Pathology Unit of Hospital Dr. José Molina Orosa for their invaluable help diagnosing this case.


