

Sr. Director:
El síndrome de Edwards o trisomía 18 es una cromosomopatía que presenta una gran variedad de manifestaciones clínicas. Las lesiones cutáneas, junto con otros rasgos polimalformativos, pueden orientar a su diagnóstico neonatal en aquellos casos que han pasado desapercibidos durante la gestación. Presentamos dos casos afectos de esta rara enfermedad.
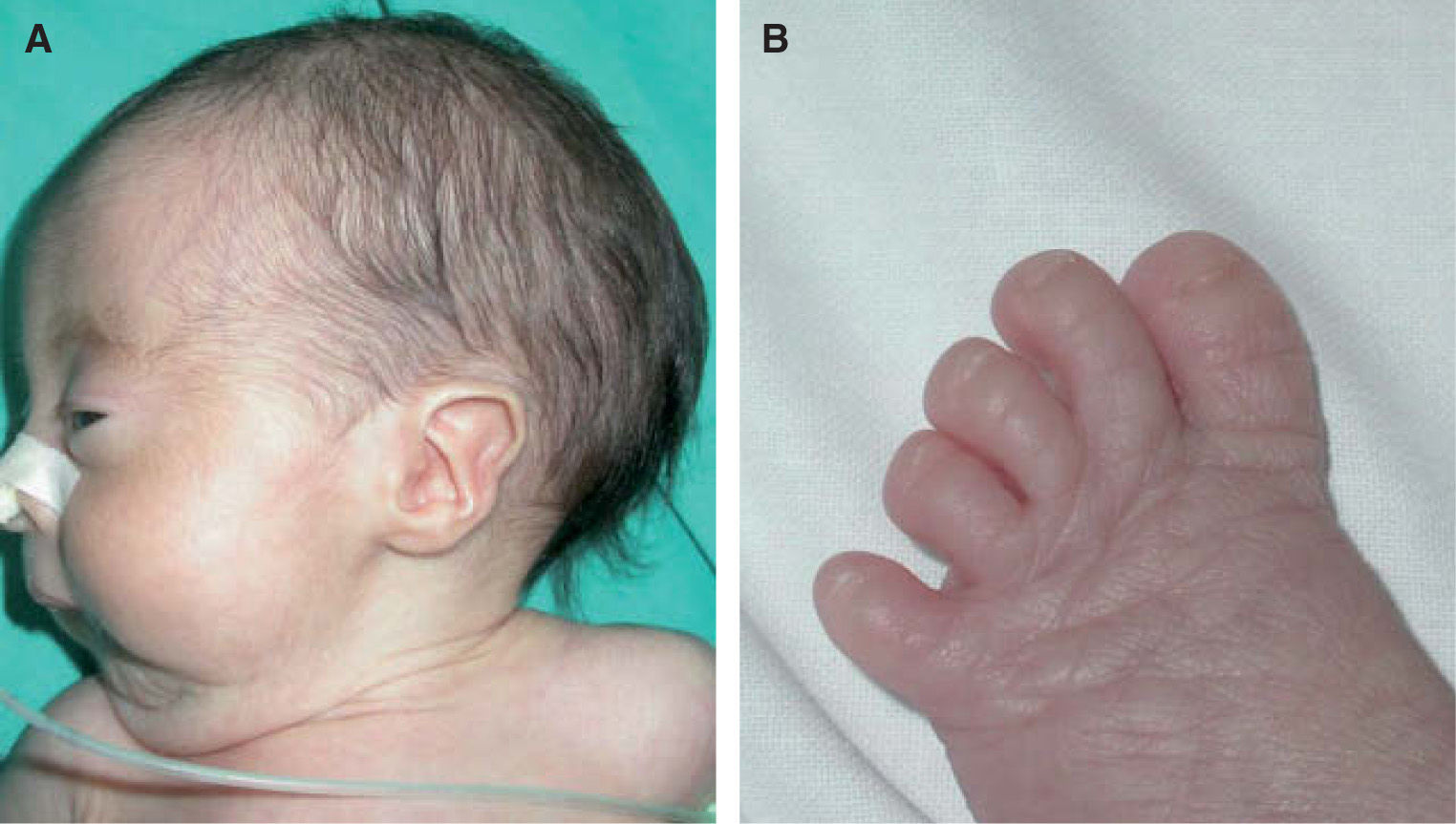
Caso 1: recién nacido a término, de madre de 42 años de edad, que en la exploración física mostraba una fascies dismórfica con orejas displásicas de implantación baja, retrognatia e hirsutismo frontal. Clínicamente también destacaba la presencia de una hipoplasia ungueal en manos y pies, una hipoplasia de labios mayores con un clítoris prominente y una sindactilia del segundo-tercer dedo del pie derecho con presencia de un calcáneo prominente (pie en mecedora). Además, los dedos de la mano adoptaban una disposición característica, el segundo montado sobre el tercero (mano trisómica) (figs. 1 y 2). Durante la gestación no se estableció el diagnóstico sindrómico, siendo informadas las ecografías gestacionales de control como normales. Ante los hallazgos clínicos se indicó la realización de un cariotipo de confirmación. El resultado del cariotipo fue 47xx+18, estableciéndose el diagnóstico definitivo de síndrome de Edwards. Las exploraciones complementarias mostraron la presencia de un ductus arterioso persistente, comunicación interauricular de tipo ostium secundum, quistes aracnoideos encefálicos y riñones en herradura. A las 7 semanas de vida presentó una insuficiencia respiratoria con edema agudo de pulmón, como complicación de la cardiopatía congénita, y falleció pocas horas después.


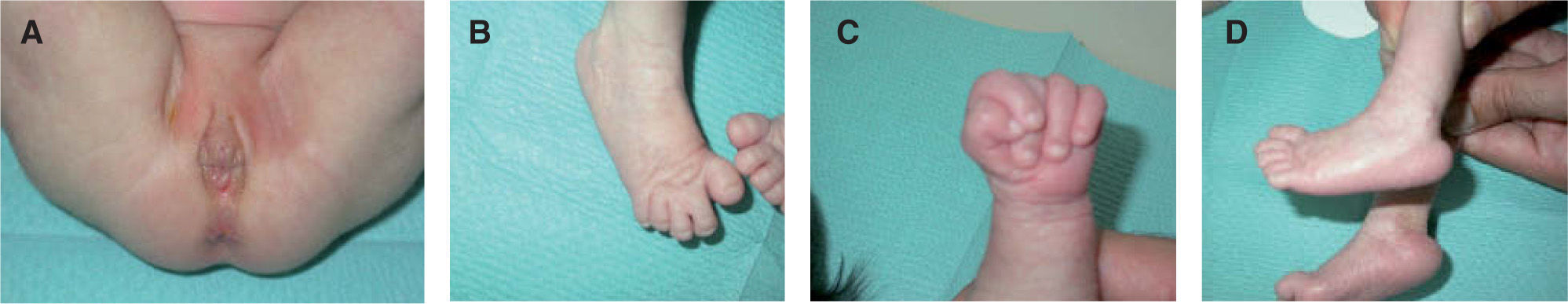
Caso 2: recién nacido a término, al que en el control ecográfico del tercer trimestre de gestación se le apreciaron múltiples defectos del tubo neural, una comunicación interventricular y una posible transposición de grandes vasos. Por dicho motivo, en la semana 34 se le realiza un estudio genético del cariotipo mediante técnica de FISH (hibridación con fluorescencia in situ), cuyo resultado fue el de un feto varón con trisomía del cromosoma 18 (47xy+18). Al nacimiento mostraba una intensa cianosis con retraso del crecimiento, un Apgar de 2 y un disrafismo espinal (mielomeningocele lumbar).También se evidenció un hirsutismo frontal, una hipoplasia ungueal de manos y pies, un patrón dermatoglífico con arcos en la totalidad de las yemas de los dedos, así como la presencia de máculas azuladas de aspecto reticulado que afectaban la piel del tronco y extremidades compatible con un cutis marmorata. El feto falleció a las 4 horas del nacimiento.
La trisomía 18, descrita por Edwards en 19601, es el segundo síndrome de malformaciones múltiples en orden de frecuencia tras la trisomía del 21. Su incidencia se estima entre 1/6.000-1/13.000 recién nacidos vivos y aparece más frecuentemente en mujeres (3:1). La edad avanzada de la madre puede ser un factor de riesgo para su desarrollo2. Presenta una alta letalidad, con una mortalidad del 90 % en el primer año de vida3,4. Las principales causas de muerte son las cardiopatías congénitas, las apneas y las neumonías5. Se han descrito más de 130 anomalías clínicas distintas en estos pacientes. Pueden presentar retraso pondo-estatural; malformaciones craneales adoptando una fascies característica con microcefalia, occipucio prominente, orejas de implantación baja y micrognatia; diversas cardiopatías congénitas objetivables en un 90 % de los casos; alteraciones urogenitales como el riñón en herradura; presencia de una mano trisómica o de un talón prominente en las extremidades, y otras múltiples malformaciones congénitas a nivel gastrointestinal y del sistema nervioso central. Entre los hallazgos cutáneos típicos se encuentran la hipoplasia ungueal de manos y pies, el hirsutismo en frente y espalda, la ausencia de panículo adiposo en el nacimiento, un patrón dermatoglífico con arcos en al menos 6 pulpejos de los dedos y lesiones reticuladas persistentes de cutis marmorata como también ocurre en el síndrome de Down, síndrome de Cornelia de Lange, hipotiroidismo congénito y lupus neonatal. El cutis marmorata persistente de estos pacientes no se debe confundir con el cutis marmorata telangiectásico congénito; este último provoca flebectasias y telangiectasias asociadas a úlceras y atrofia de extremidades2,5.
En conclusión, nos gustaría destacar la importancia de las manifestaciones cutáneas, que aparecen en más del 50 % de los pacientes con síndrome de Edwards, porque asociadas con otros rasgos polimalformativos sirven de orientación diagnóstica. El diagnóstico definitivo se establece tras la realización de un cariotipo, que confirma la existencia de la trisomía 181,5,6.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.



