


La incontinencia pigmenti (síndrome de Bloch-Sulzberger) es una displasia neuroectodérmica infrecuente, con patrón de herencia ligado al X dominante, causada por mutaciones en el gen IKBKG/NEMO y se encuentra localizado en Xq28. La deleción del exón 4 al 10 corresponde con la principal causa en aproximadamente el 80% de los casos. La incidencia estimada es de 0,7 en 100.000 nacimientos, usualmente letal en hombres hemicigotos y en el sexo femenino puede exhibir hallazgos clínicos variables. Es una entidad multisistémica, que incluye defectos en la piel, siempre presente y principal criterio diagnóstico, la cual evoluciona en cuatro etapas, asociada a alteraciones en el sistema nervioso central, globo ocular, dientes, glándula mamaria, pelo, uñas, entre otros. El objeto de esta breve revisión es resaltar los hallazgos clínicos de esta genodermatosis, con la finalidad de brindar el seguimiento médico individualizado e interdisciplinario que incluya un adecuado asesoramiento genético familiar.
Incontinentia pigmenti (Bloch-Sulzberger syndrome) is a rare neuroectodermal dysplasia. It is an X-linked dominant disorder caused by mutations in the IKBKG/NEMO gene on Xq28. Approximately 80% of patients have a deletion of exons 4 to 10. Incontinentia pigmenti has an estimated incidence of 0.7 cases per 100,000 births. In hemizygous males, it is usually lethal, while in females, it has a wide spectrum of clinical manifestations. Incontinentia pigmenti is a multisystemic disease that invariably features skin changes. These changes are the main diagnostic criteria and they evolve in 4 stages, in association with other abnormalities affecting the central nervous system, eyes, teeth, mammary glands, hair, nails, skin, and other parts of the body. The aim of this brief review is to highlight the clinical features of this genodermatosis and underline the importance of case-by-case interdisciplinary management, including genetic counseling.
La incontinencia pigmenti (IP, OMIM 308300) es una displasia neuroectodérmica infrecuente1,2, con patrón de herencia ligado al X dominante, causada por mutaciones en el gen IKBKG/NEMO (GenBankNM_003639.3, OMIM 300248), anteriormente llamado NEMO y localizado en Xq28. Codifica un inhibidor del potenciador del gen del polipéptido ligero kappa en células B Cinasa Gamma, el cual es un modulador esencial del factor de transcripción nuclear kappa B1,3–7.
Fue descrita por Bloch en 1926, y Sulzberger en 19284, por lo que también es conocida como el síndrome de Bloch-Sulzberger6,8. Presenta alta penetrancia y expresividad variable4. La deleción de 11,7kb del exón 4 al 10 corresponde la principal causa, en aproximadamente el 80% de los casos3,6,9,10. Es causada por la recombinación entre dos repeticiones MER67B localizadas en los intrones 3 y 10 del gen, con consecuente abolición de la función de la proteína. Adicionalmente, pequeñas mutaciones de tipo inserción-deleción o sin sentido han sido descritas. Algunas mutaciones hipomórficas del gen IKBKG/NEMO causan una entidad distintiva denominada displasia ectodérmica hipohidrótica con inmunodeficiencia (OMIM 300291)6,11.
EpidemiologíaLa incidencia estimada es de 0,7 en 100.000 nacimientos10, con 27,6 casos nuevos por año en el mundo. Entre 65 a 75% se deben a mutaciones esporádicas y entre 25 a 35% son casos familiares2. Es usualmente letal en período embrionario en hombres hemicigotos2–4,12,13, y el sexo femenino puede sobrevivir debido al mosaicismo funcional en la inactivación del cromosoma X2,4,9, con hallazgos clínicos altamente variables3. Por lo tanto, es predominante en el sexo femenino con una relación 37:1. En un individuo masculino con hallazgos clínicos de IP puede coexistir el síndrome de Klinefelter (47, XXY), en el cual el segundo cromosoma X puede permitir la supervivencia o en casos de mosaicismo somático para la deleción común, ya comentada4,5,8.
Clínica y manejoLa IP es una entidad multisistémica4,5,8, que incluye los tejidos de origen ectodérmico y mesodérmico4. Afecta a la piel, la cual siempre se encuentra presente y corresponde el principal criterio diagnóstico9. Se encuentra igualmente asociada a alteraciones en sistema nervioso central, globo ocular, dientes3,4,10,12, glándula mamaria, pelo, uñas, anomalías esqueléticas, entre otras menos frecuentes como las anormalidades cardiopulmonares12.
En 1993, se determinaron los criterios diagnósticos originales por Landy y Donnai14, antes del descubrimiento del gen causante10. En 2014, Minić et al.15. propusieron actualizaciones y posteriormente en 2018, Rosser3, modificó con permiso de Minić et al., estos criterios con el objeto de mejorar la caracterización fenotípica y proponer una actualización en el diagnóstico. Los criterios mayores describen los hallazgos dermatológicos típicos y asociados a la entidad, que realizan el diagnóstico clínico3,5, junto a los criterios menores asociados a otras anomalías que pueden evidenciarse. Se han propuesto condiciones adicionales para establecer el diagnóstico como la presencia de un familiar de primer grado de sexo femenino y la mutación en el gen IKBKG/NEMO (tabla 1)2–20, en la que se describen los criterios citados por Rosser y en cada uno de ellos se mencionan los hallazgos encontrados en la literatura.
Criterios propuestos para el diagnóstico de la IP y porcentajes de algunos de ellos según lo encontrado en la literatura
| Criterios | Frecuencia (%) |
|---|---|
| Mayores | |
| Lesiones típicas en la piel que se distribuyen en las líneas de Blaschko | 100 |
| (Etapa I) vesicobulosa: lesiones eritematosas o inflamatorias, caracterizadas por vesículas, bulas o pústulas. Más frecuente en extremidades y cuero cabelludo | |
| Presente desde el nacimiento hasta la segunda semana de vida | |
| (Etapa II) verrugosa/hiperqueratósica: pústulas o costras, hiperpigmentadas | |
| Ocurre desde la segunda a la sexta semana | |
| (Etapa III) hiperpigmentada: máculas hipercrómicas en forma de espiral. Predominan en áreas intertriginosas y tronco | |
| Aparecen entre 12 a 26 semanas y pueden mejorar en la adolescencia o persistir en la edad adulta. | |
| (Etapa IV) atrófica/hipopigmentada: máculas hipocrómicas, puede presentar alopecia | |
| Esta etapa no está presente en todos los casos | |
| Estas etapas pueden superponerse y algunas veces seguir de forma irregular | |
| Menores | |
| Anomalías en sistema nervioso central/neurológicas | 30 |
| Convulsiones | 40,2 |
| Parálisis espástica | 16,5 |
| Retraso psicomotor | 26 |
| Retraso mental | 29,9 |
| Microcefalia | 11,3 |
| Atrofia cerebral/cerebelar | 13,4* |
| Microgiria/polimicrogiria | |
| Hipoplasia en cuerpo calloso | |
| Alteración en ganglios de la base | |
| Leucomalacia periventricular | |
| Hidrocefalia | 5,1 |
| Porencefalia | |
| Accidente cerebrovascular isquémico | |
| Necrosis hemorrágica difusa | |
| Encefalomielitis | |
| Anomalías oculares | |
| Defectos en la visión | 17 |
| Retinopatía | 15,9 |
| Desprendimiento de retina | 8,5 |
| Alteración vascular en retina | 5,3 |
| Anormalidades pigmentarias hiper/hipopigmentado | |
| Atrofia óptica | |
| Hipoplasia foveal | |
| Fibroplasia retrolental | |
| Cataratas | |
| Microftalmia | 6,4 |
| Estrabismo | |
| Nistagmo | |
| Anomalías dentales | 17-34 |
| Retardo en la erupción primaria | 34,3 |
| Anodoncia/hipodoncia | |
| Microdoncia | |
| Distrofia dental | 17,2 |
| Anomalías de la forma (cónicos) | 22,3 |
| Impactación | 17,2 |
| Diastema | |
| Maloclusión | |
| Alteraciones en paladar | |
| Paladar alto | |
| Alteraciones glándula mamaria | |
| Pezón supernumerario | |
| Anormalidades en pelo (pelo, cejas, pestañas) | 28-38 |
| Alopecia | 9,7 |
| Hipertricosis | 3,6 |
| Anormalidades en uñas | |
| Distrofia | 64 |
| Pigmentación amarillenta | |
| Hendiduras transversales o longitudinales | |
| Abortos de fetos de sexo masculino | |
| Hallazgos histopatológicos típicos en piel | |
| Condiciones adicionales que confirman el diagnóstico | |
| No existe evidencia de IP en familiar femenino de primer grado | |
| Si no hay disponibilidad de realizar estudio molecular, se requieren dos o más criterios mayores o un criterio mayor y uno menor para confirmar el diagnóstico | |
| Mutación en IKBKG con cualquier criterio mayor o menor confirma el diagnóstico | |
| Evidencia de IP en un familiar femenino de primer grado | |
| Necesita un criterio mayor o dos criterios menores | |
| Eosinofilia e inactivación del cromosoma X sesgada apoyan el diagnóstico en todos los casos |
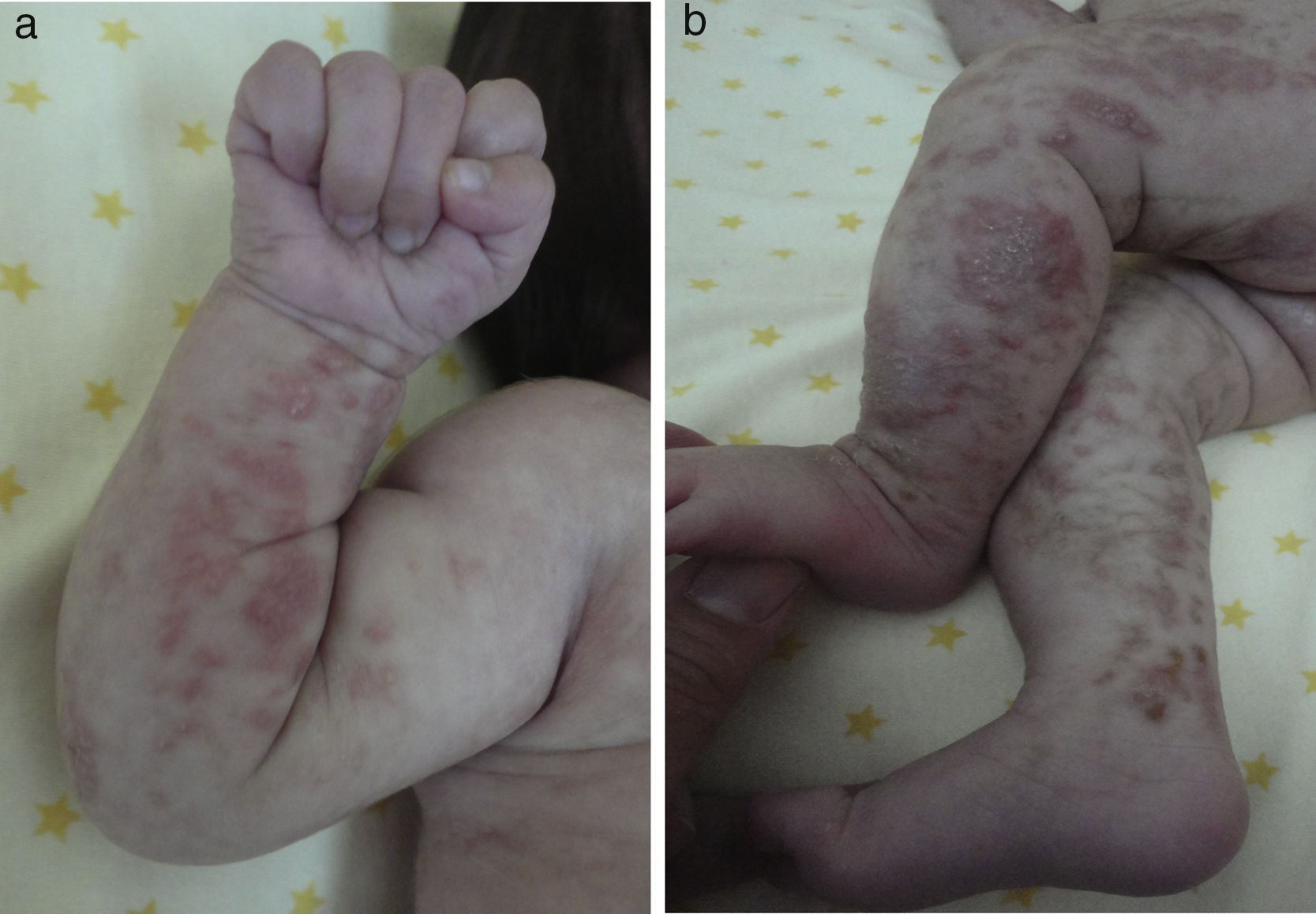
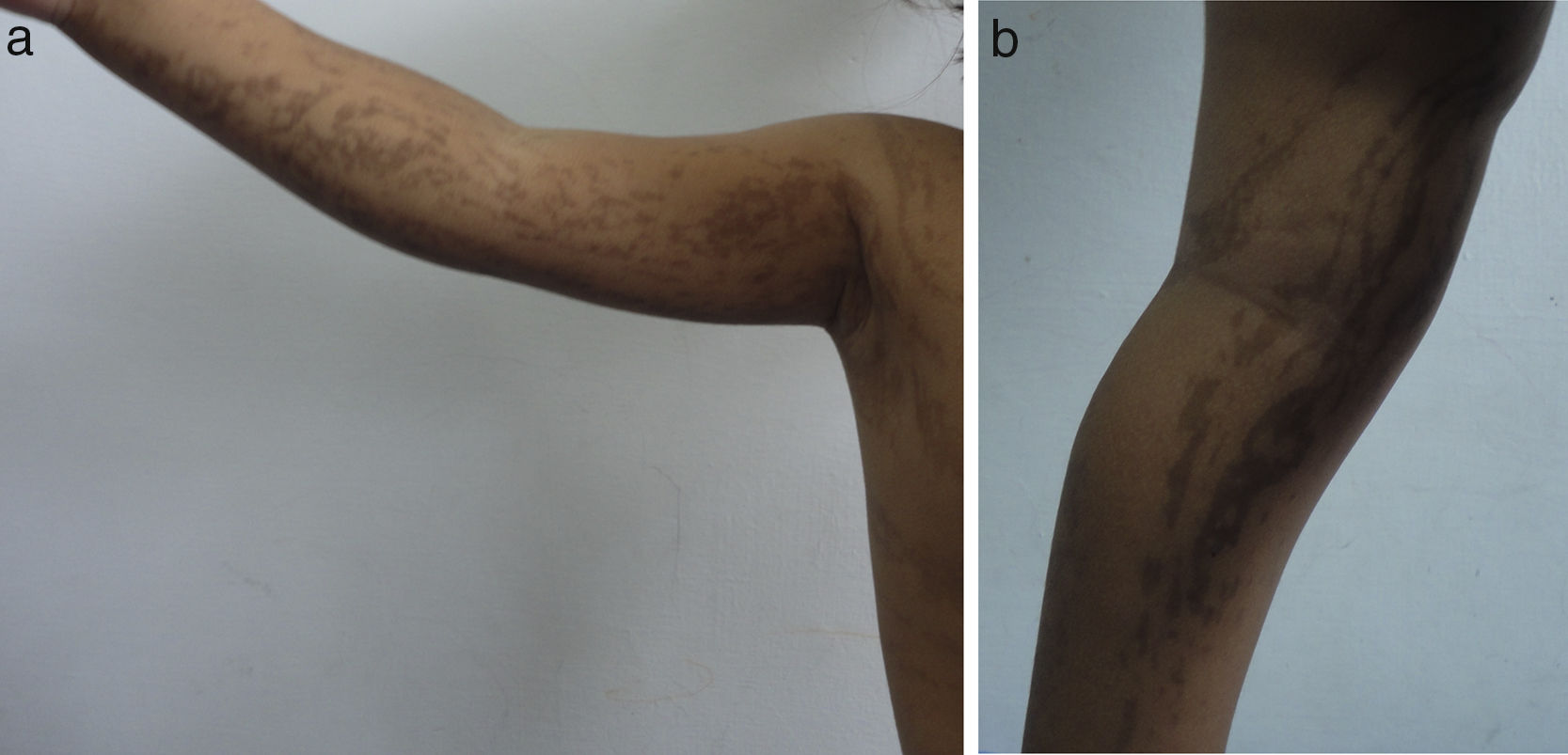
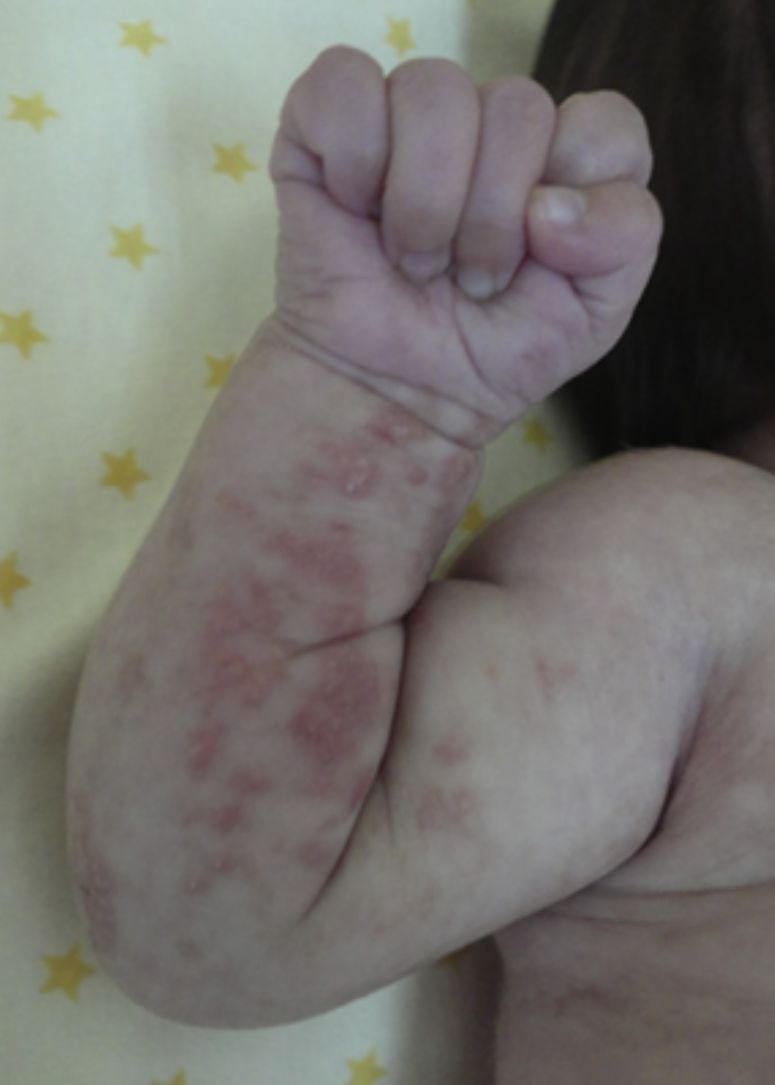
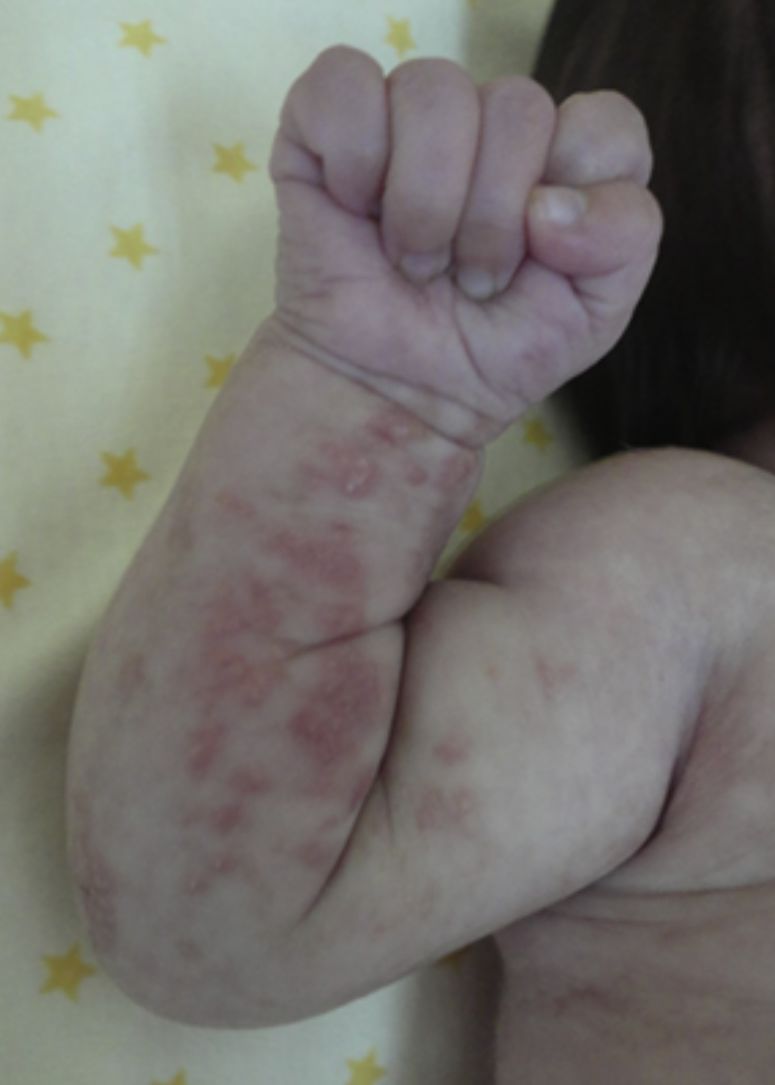
Las lesiones en piel corresponden a la primera manifestación clínica, comienza al momento del nacimiento o en las primeras dos semanas de vida, evolucionan por años, se encuentran dispuestas a lo largo de las líneas de Blaschko y se presentan en cuatro etapas clásicas: vesicobulosa (fig. 1), verrugosa/hiperqueratósica, hiperpigmentada (fig. 2), y atrófica/hipopigmentada19,20. En algunos casos, no todas las lesiones se presentan19, y las tres primeras etapas pueden aparecer de forma simultánea20. Estas lesiones se describen más ampliamente en la (tabla 1)2–20. El nombre de IP está relacionado con las características histológicas de las lesiones en la tercera etapa (hiperpigmentada) que consiste en la incontinencia por los melanocitos de la capa epidérmica basal y por su presencia en la dermis superficial4. Estas manifestaciones generalmente no requieren de intervención3, y no amerita un tratamiento específico. El tratamiento con corticosteroides tópicos o tacrolimus puede detener la progresión de la fase vesicular, aunque las lesiones desaparecen espontáneamente18.


La gravedad de la entidad se encuentra principalmente relacionada con la presencia de alteraciones neurológicas9,10, que puede conducir a una importante morbimortalidad19. Estas anormalidades son variadas e incluyen a las convulsiones como el hallazgo más común9,21, por lo general de tipo tónico22, y clónico focal, de corta duración con pérdida del estado de conciencia, siendo más frecuente en el período neonatal9, resistente al tratamiento22, y considerado de mal pronóstico. Además, se puede evidenciar microcefalia, accidente cerebrovascular isquémico, ataxia cerebelosa, y retraso psicomotor, entre otros. Los signos agudos de la encefalitis ocurren principalmente en los primeros cuatro días de vida e incluyen intolerancia a la alimentación, letargia y movimientos similares a las convulsiones. Por lo tanto, se debe realizar resonancia magnética cerebral con angiograma para detectar las posibles alteraciones neurológicas desde el período neonatal en pacientes con lesiones cutáneas características de la IP, con posteriores controles, para así evitar las secuelas que pueden presentarse3,9,22.
Las anormalidades oculares se han encontrado en el 50 a 77% de los pacientes. Las alteraciones en retina son las más importantes16, y cuyas complicaciones son las más temidas, como el desprendimiento de retina y la retina avascular, los cuales se pueden tratar con fotocoagulación con láser, con respuesta para la neovascularización retiniana. La crioterapia también puede ser utilizada y los inhibidores del factor de crecimiento endotelial vascular parecen ser prometedores18. Por su parte, las imágenes en tomografía de coherencia óptica muestran el adelgazamiento de las capas de la retina interna y externa. Se han descrito anomalías oculares similares al retinoblastoma en algunos casos de IP, como la leucocoria, estrabismo, calcificación intraocular y el ya comentado desprendimiento de retina, por lo que es necesario establecer el diagnóstico diferencial16. Se recomienda que la evaluación ocular debe ser mensual durante los primeros cuatro meses, cada tres meses hasta el primer año de edad, dos veces al año hasta los 3 años, y luego anualmente18.
EtiopatogeniaEl factor de transcripción nuclear kappa B es activado por la proteína producto del gen IKBKG/NEMO, esencial modulador que desempeña un papel importante en múltiples funciones fisiológicas como la respuesta inmune y el estrés, las reacciones inflamatorias, el desarrollo ectodérmico, la adhesión celular y la protección de las células contra apoptosis inducida por el factor de necrosis tumoral3,4,6,22.
Por lo tanto, la mutación en el gen IKBKG/NEMO, conduce a la disminución de la actividad del factor de transcripción nuclear kappa B, lo cual aumenta la susceptibilidad de las células a la apoptosis4. Por ello, en el sexo masculino la extensa apoptosis es responsable de la letalidad fetal temprana10, así como alteraciones a nivel hepático2.
Además, las reacciones inflamatorias y el reclutamiento epidérmico de eosinófilos, observado en la primera etapa de la entidad, suele ser importante en su patogenia4. Es probable que esté relacionado con una quimiocina selectiva de eosinófilos (eotaxina)4,16, producida por leucocitos específicos como los eosinófilos, macrófagos y células T, así como algunas células estructurales como las endoteliales, los fibroblastos y las epiteliales4.
Diagnóstico diferencialVaría de acuerdo con la etapa de la entidad, en la primera puede confundirse con múltiples infecciones del recién nacido como el herpes simple congénito, varicela, además de la epidermólisis ampollar o el penfigoide ampolloso18,23. Dado que el herpes neonatal y la IP pueden coexistir, se debe iniciar el tratamiento temprano con aciclovir, si se confirma el diagnóstico de la infección viral. En la segunda etapa es limitado e incluye el nevo epidérmico lineal23. La tercera etapa es el sello distintivo de la IP y se debe diferenciar con la hipermelanosis nevoide lineal o en espiral. Por su parte, la cuarta etapa de estar presente, se puede diagnosticar erróneamente como hipomelanosis de Ito o vitíligo. Tanto la hipermelanosis nevoide lineal o espiral y la hipomelanosis de Ito carecen de una fase inflamatoria inicial y generalmente no se encuentran comprometidos los anejos18.
Conclusiones y asesoramiento genéticoEl diagnóstico de la IP es clínico18. No obstante, la gran heterogeneidad clínica y el espectro de alteraciones moleculares dificultan la selección de un grupo homogéneo de pacientes, que puede dificultar la estandarización de un abordaje terapéutico. A pesar del progreso en describir la etiología del trastorno y las amplias investigaciones en la atención clínica, la escasez de pacientes en los centros de diagnóstico hace difícil un informe epidemiológico global. La integración de estos datos puede ser crucial para el éxito de logros científicos futuros10.
Ante un trastorno multisistémico, el seguimiento médico debe realizarse a largo plazo y de forma individualizada e interdisciplinaria comprendiendo las evaluaciones por los servicios de Pediatría, Dermatología, Neurología, Oftalmología y Odontología entre otras diversas especialidades3,24. Se ha descrito hipotiroidismo congénito, miastenia gravis y tumor de Wilms en algunos casos de IP18, e incluso aunque no se puede establecer el vínculo, recientemente se sugiere la evaluación hepática1, por lo que se amplía aún más la atención médica ante un paciente con esta entidad.
Por otro lado, la actualización en el diagnóstico propuesta por Rosser3, no incluye las alteraciones esqueléticas entre ellas, la talla baja, hemivértebra, cifosis, escoliosis, clavículas supernumerarias, displasia de cadera, hemiatrofia, pie equinovaro4, y sindactilia en dedos de los pies4,10, así como los trastornos cardiopulmonares, como la comunicación inteauricular4, fibrosis endomiocárdica ventricular izquierda, insuficiencia tricuspídea, tetralogía de Fallot4,17, y la hipertensión pulmonar incluso en ausencia de alteración cardiovascular2,4. Adicionalmente, el riesgo de contraer infecciones recurrentes10, tampoco se encuentra citado en estos criterios. Es probable que futuras actualizaciones se describan, con el objeto de ampliar las principales características clínicas que pueden estar presentes.
La evaluación por Genética es crucial para facilitar la comprensión de la entidad y permite confirmarla a través del estudio genético molecular24, útil en casos dudosos18. El pronóstico es generalmente bueno, pero se debe realizar como ya se ha hecho referencia, la evaluación periódica por parte del equipo interdisciplinario. El asesoramiento genético familiar es igualmente importante18,23, ante la presencia de un patrón de herencia ligado al X dominante usualmente letal en hombres, una paciente femenina con la entidad tiene un riesgo de 50% de tener hijas con IP. Ante un feto de sexo masculino se puede presentar aborto en un 50% y de presentarse un hijo afectado está indicada la realización del estudio citogenético.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nuestro agradecimiento a Incontinentia Pigmenti Genetic Biobank (www. igb. cnr. it/ ipgb) del Biobanking and BioMolecular Resources Research Infrastructure-European Reasearch Infrastructure Consortium (BBRMI-ERIC) y CNR-DSB Proyecto Bandera «InterOmics».
A la licenciada Rosalía Gumina F., directora de la Biblioteca del Instituto Autónomo Hospital Universitario de Los Andes, Universidad de Los Andes.



