


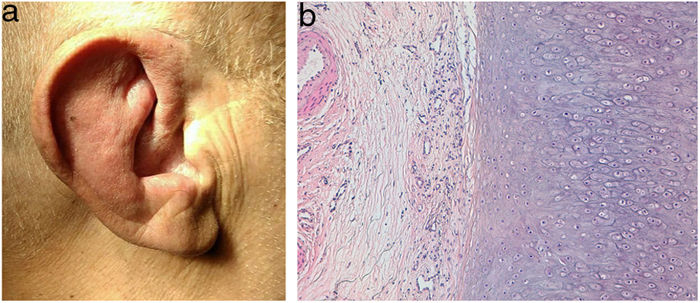
Un hombre de 71 años presentaba una inflamación recurrente de los pabellones auriculares (fig. 1a) de 6 meses de evolución, por lo que había sido diagnosticado de policondritis recidivante. Además, presentaba unas lesiones nodulares eritematosas en las extremidades y el tronco.
 Eritema y edema de los pabellones auriculares. b) Biopsia de pabellón auricular, mostrando cartílago degenerado e inflamación crónica en pericondrio. Hematoxilina-eosina, ×100.")
El paciente refería una pérdida de 21 kg de peso en el último año, sin fiebre asociada. Entre los problemas médicos concomitantes destacaba una epiescleritis bilateral, una fibrilación auricular, una epididimitis de repetición y una pancitopenia.
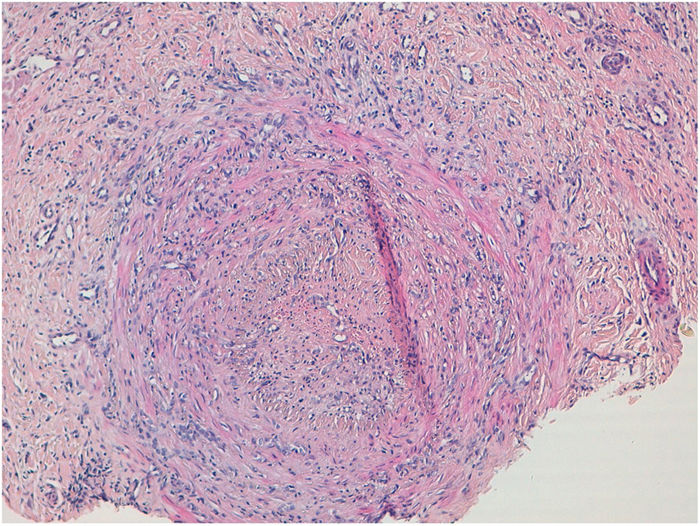
HistopatologíaEn el estudio histológico del cartílago auricular se observó una infiltración linfocitaria en el pericondrio (fig. 1b), mientras que en la biopsia de las lesiones nodulares cutáneas se objetivó una vasculitis de arteria de mediano calibre (fig. 2), con un infiltrado inflamatorio en la pared vascular predominantemente neutrofílico y oclusión de la luz vascular.
Otras pruebas complementarias
Los reactantes de fase aguda se encontraban elevados, presentando una ferritina de 6.830 (VR: 20-250) y una PCR de 130 (VR: 0-5).
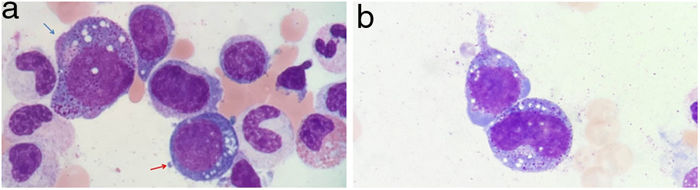
Se realizó un aspirado de médula ósea para el estudio de la pancitopenia, revelando una displasia multilínea y una llamativa vacuolización citoplasmática y nuclear de la serie mieloide y eritroide (fig. 3a y b).
 Vacuolización de núcleo y citoplasma de precursores mieloides (flecha azul) y eritroides (flecha roja) en aspirado medular. May-Grunwald-Giemsa, ×100. b) Vacuolización de núcleo y citoplasma de precursores mieloides medulares. MayGrunwald-Giemsa, ×100.")
Para la confirmación diagnóstica del síndrome sospechado, se solicitó estudio del gen UBA1, encontrándose la mutación c.122T>C, p.(Met41Thr).
¿Cuál es el diagnóstico?
DiagnósticoSíndrome VEXAS (del inglés vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic).
Tratamiento y evoluciónEl paciente recibió tratamiento fuera de ficha técnica con adalimumab, falleciendo a los 5meses del diagnóstico.
ComentarioEl síndrome VEXAS es un cuadro autoinflamatorio de inicio en la edad adulta, descrito en diciembre del 2020 por Beck et al.1. Se debe a una mutación somática en el gen UBA1, que ocasiona una ubiquitinización disfuncional y, consecuentemente, un estrés proteotóxico con activación de la inmunidad innata. Se ha descrito principalmente en varones de mediana edad2.
Los pacientes desarrollan signos y síntomas sistémicos, como fiebre, astenia y pérdida de peso, presentando lesiones cutáneas hasta en el 83% de los casos2, con frecuencia en forma de pápulas y nódulos eritemato-violáceos. El patrón histológico predominante en este tipo de lesiones es el de una infiltración neutrofílica, con o sin vasculitis. En la serie de Beck et al., el 32% de los pacientes habían sido diagnosticados previamente de síndrome de Sweet y el 12% de poliarteritis nudosa1. No obstante, también se han descrito casos en los que en la biopsia cutánea se evidenció un infiltrado perivascular linfocitario, sin neutrófilos ni vasculitis3. Otros hallazgos cutáneos descritos en esta entidad son la livedo racemosa y la policondritis recidivante, observándose esta última hasta en el 64% de los pacientes4.
Desde el punto de vista hematológico es muy característico la presencia de citopenias, síndrome mielodisplásico y vacuolización de los precursores mieloides y eritroides en la biopsia de médula ósea1. No obstante, la vacuolización de los precursores no es patognomónica de esta entidad, pues también se puede encontrar en pacientes alcohólicos, con un déficit de cobre o debido a la propia neoplasia de la serie mieloide.
Otras manifestaciones clínicas descritas en este síndrome son artritis, serositis, escleritis y epiescleritis, pericarditis, miocarditis, alveolitis neutrofílica, epididimitis, vasculitis de grandes vasos, tromboembolia venosa, dolor abdominal y hemorragia digestiva, entre otras2. En los análisis, se observa una elevación de los reactantes de fase aguda.
La enfermedad suele tener un curso agresivo. Sin embargo, dado que esta entidad se ha descrito recientemente, debemos tomar estos datos con cautela, ya que es posible que los casos más indolentes hayan pasado inadvertidos y el estudio genético se haya realizado en los pacientes con manifestaciones más graves. Recientemente se ha observado que los pacientes con la mutación p.Met41Leu en UBA1 podrían tener un mejor pronóstico2.
El tratamiento de este síndrome todavía no se ha consensuado. Se ha comunicado el empleo de diferentes fármacos, entre los que figuran corticoides sistémicos, azatioprina, metotrexato, micofenolato, ciclofosfamida, anti IL-1, anti IL-6, anti-TNF, rituximab, inmunoglobulinas intravenosas, abatacept y anti-JAK, obteniéndose resultados variables5. Recientemente, se ha propuesto el trasplante alogénico de precursores hematopoyéticos como una opción con resultados prometedores y mantenidos en el tiempo6.
El síndrome VEXAS es una entidad compleja, de reciente descripción, y evolución fatal en la mayoría de los casos, por lo que consideramos de interés su conocimiento con el fin de favorecer el diagnóstico y el tratamiento precoces. En los próximos años es posible que se pueda desarrollar un tratamiento molecular dirigido a restaurar la ubiquitinización de estos pacientes.
FinanciaciónNinguna.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



