




Epidermolysis bullosa (EB) is a rare genetic disease that causes mucocutaneous fragility. It comprises a clinically and genetically heterogeneous group of disorder characterized by spontaneous or contact/friction–induced blistering. EB is classified into 4 types–simplex, junctional, dystrophic, and Kindler syndrome–and 30 subtypes. The disease is caused by defects in proteins implicated in dermal-epidermal adhesion. At least 19 genes have been characterized and more than 1000 mutations identified, thus rendering diagnosis complex. Molecular diagnosis of EB is the last stage of a laborious process that starts with a detailed clinical history compilation and careful procurement of a skin fresh biopsy that includes an area where the epidermis detaches from the dermis. The detachment area makes it possible to establish the cleavage plane by antigen mapping and, in the best scenario, to identify a single candidate gene to search for pathogenic mutations. The results of the molecular diagnosis enable the physician to provide appropriate genetic counseling (inheritance pattern, risk of recurrence, and options for prenatal and preimplantation diagnosis) and implement subsequent preventive programs, as well as to establish a reasonable clinical prognosis facilitating access to specific therapy and rehabilitation. Lastly, molecular diagnosis is essential for the participation of patients in clinical trials, a critical issue given the current incurable status of EB. The present guidelines aim to disseminate the procedure for diagnosing EB in our laboratory and thus avoid suboptimal or incomplete clinical diagnoses. The recommendations we provide are the result of more than 10 years’ experience in the molecular diagnosis of EB in Spain.
La epidermólisis bullosa (EB), enfermedad genética de fragilidad mucocutánea rara y devastadora, es clínica y genéticamente heterogénea. Se caracteriza por la aparición de ampollas inducidas por contacto/fricción o de forma espontánea. La EB se clasifica en 4 tipos: simple, juntural, distrófica y síndrome de Kindler y en 30 subtipos. Esta genodermatosis está causada por defectos en proteínas implicadas en la adhesión dermoepidérmica, con al menos 19 genes caracterizados hasta el momento y más de 1.000 mutaciones identificadas, que explican la complejidad de su diagnóstico. El diagnóstico molecular de la EB es el último paso de un proceso laborioso que se inicia con la recogida de una historia clínica detallada y la toma de una biopsia cutánea, que incluya una zona de despegamiento entre la dermis y la epidermis inducida, en el momento de la recolección. Dicho despegamiento permite establecer el plano de rotura por mapeo antigénico y, en el mejor de los casos, un único gen candidato en el que realizar la búsqueda de las mutaciones patogénicas. Finalizado el diagnóstico molecular, se está en condiciones de ofrecer al paciente un asesoramiento genético adecuado (patrón de herencia, riesgo de recurrencia y opciones de diagnóstico prenatal y preimplantacional) y los consecuentes programas preventivos, así como un pronóstico clínico razonable que facilite su acceso a opciones terapéuticas y de rehabilitación específicas. Por último, el diagnóstico molecular es imprescindible para la participación de los pacientes en ensayos clínicos, de gran importancia en una enfermedad como la EB, que no tiene cura. El objetivo de la presente guía es difundir el procedimiento de diagnóstico de la EB tal y como se está llevando a cabo en nuestro laboratorio y, así, evitar diagnósticos clínicos subóptimos o incompletos. Las recomendaciones recogidas son fruto de nuestra experiencia de más de 10 años de diagnóstico molecular de EB en España.
The term epidermolysis bullosa (EB), often known as butterfly skin, refers to a genetically and clinically heterogeneous group of rare diseases, characterized by muco-cutaneous fragility. The disorder leads to the formation of blisters and erosions that may arise due to friction or even spontaenously.1,2 Other complications associated with EB are the appearance of hypoplastic lesions of dental enamel; airway, gastrointestinal, and urogenital stenosis or narrowing; pyloric atresia; muscular dystrophy; and cancer.2–4 Therefore, management of patients with EB requires a multidisciplinary team (for further information, see Guide for Integrated Care of Hereditary Epidermolysis Bullosa published by the Spanish Ministry of Health and Consumer Affairs5).
EB affects people of all ethnic origins and both sexes equally. The overall incidence in the United States is estimated to be 1/53 000 and the prevalence to be 1/125 000. Similar estimates have been reported in some European countries, including Spain6 (1/166 000; www.orpha.net).
EB is classed as an orphan disease; as there is no specific and effective treatment, current treatments aim merely to alleviate symptoms.7 The scientific community is going to great lengths to offer innovative solutions based on so-called advanced therapies: cell therapy, tissue genetics and engineering, and protein therapy.8–12
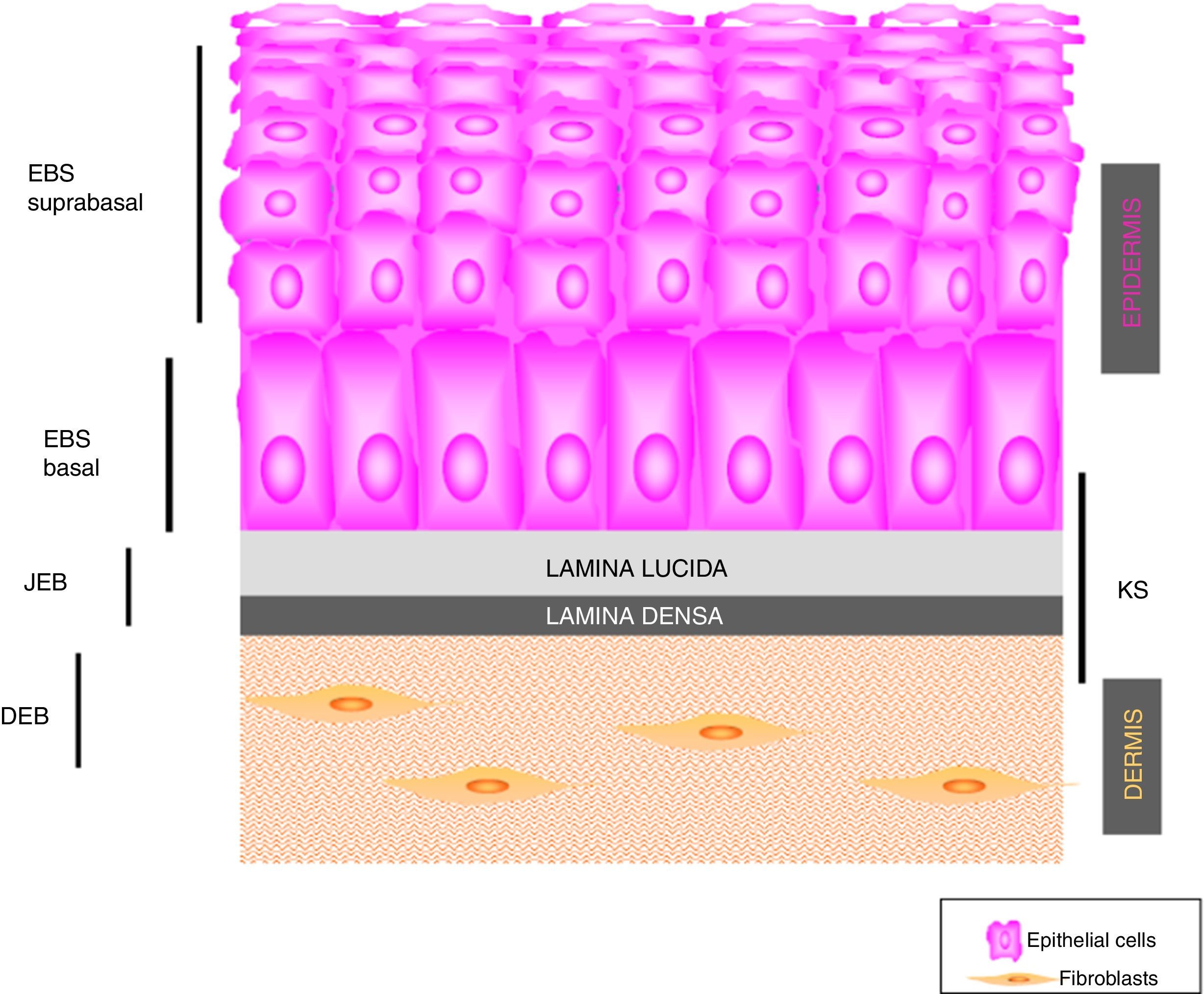
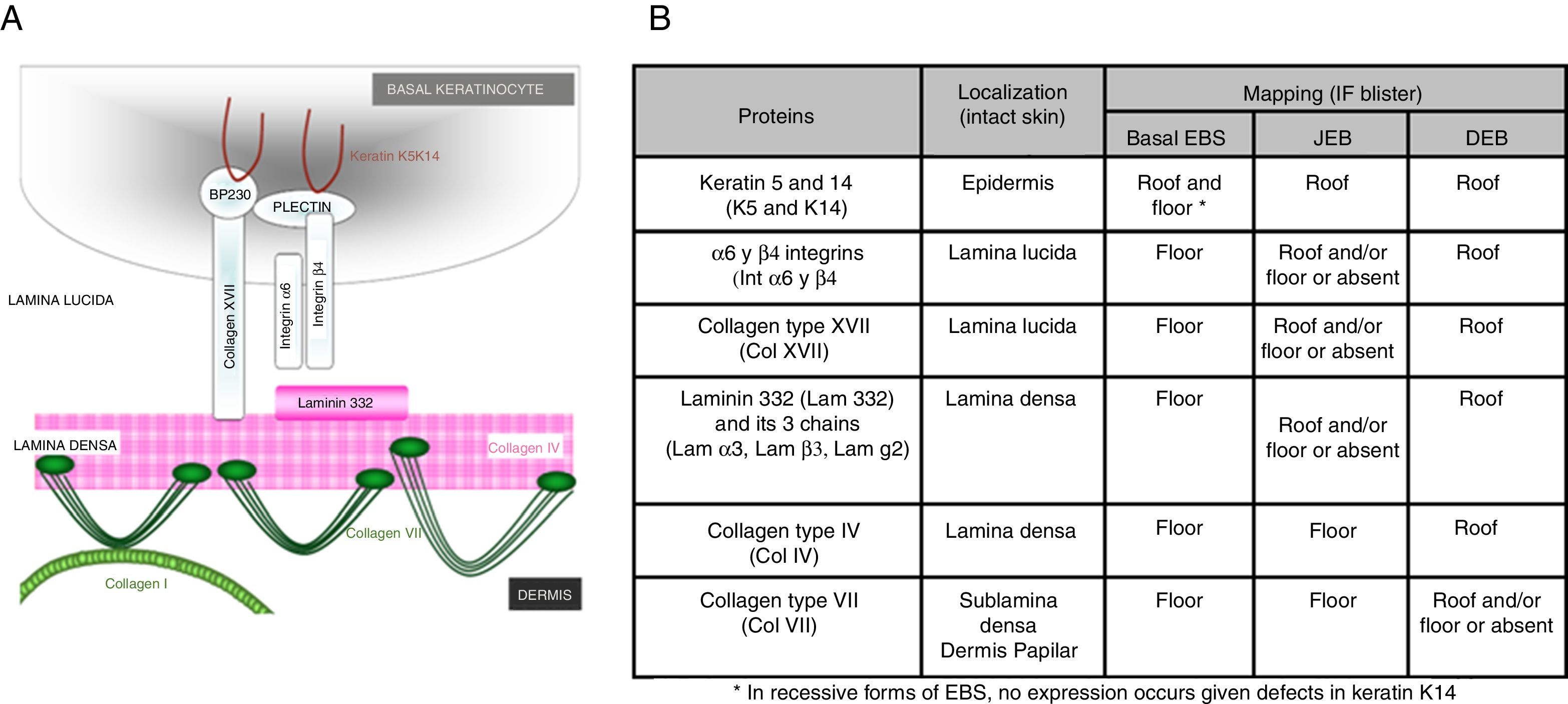
Main Types of Epidermolysis BullosaFour types of EB are defined according to the level of cleavage when a blister develops: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler syndrome (KS). In EBS, fragility is detected at an intraepidermal level (and can involve both basal or suprabasal keratinocytes). In JEB, blister formation occurs in the lamina lucida while in DEB it occurs below the lamina densa (in the papillary dermis). In KS, cleavage can occur at any level except in suprabasal layers2(Fig. 1).
Pathogenesis
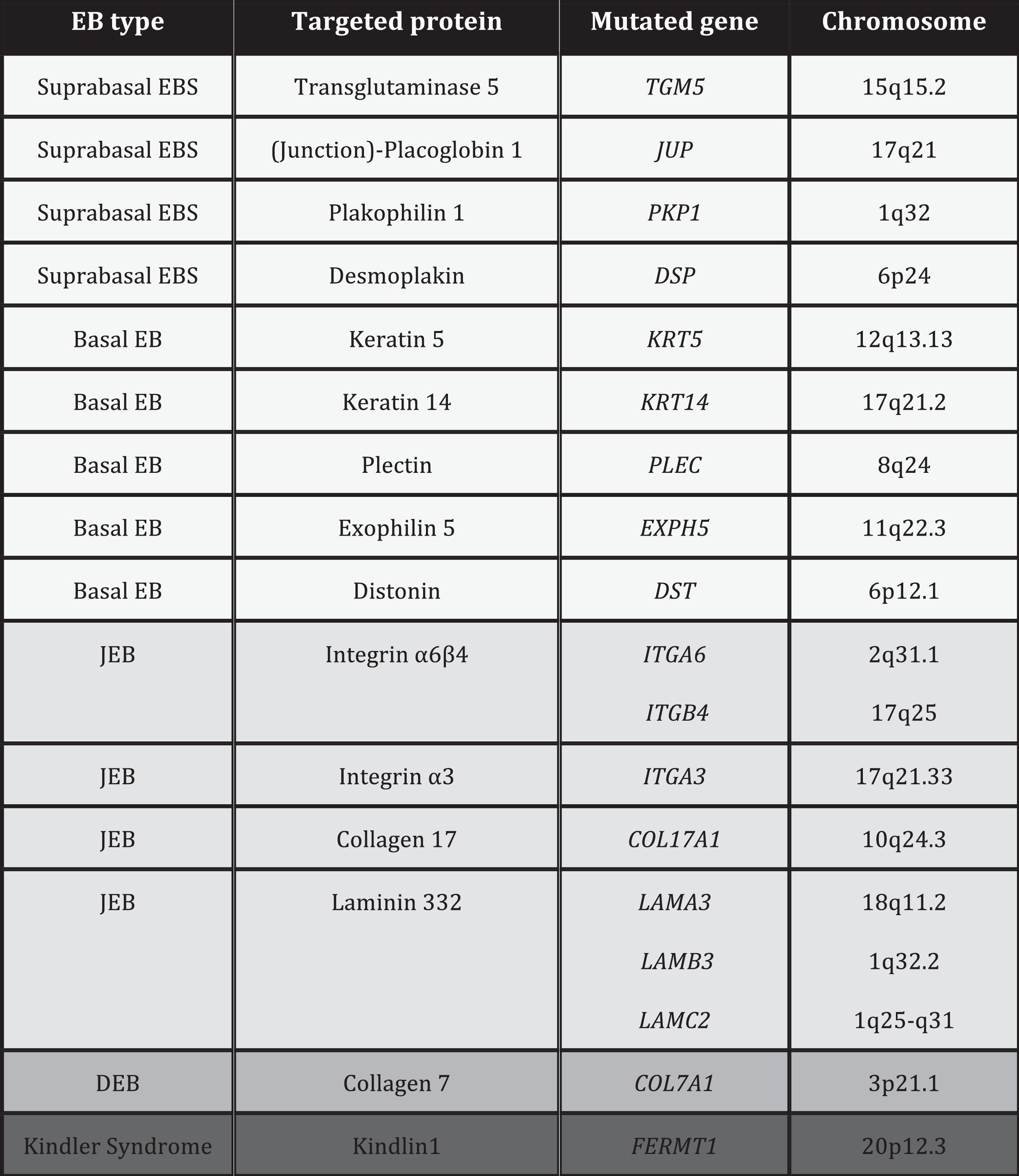
EB is caused by mutations in genes that encode proteins responsible for the integrity and mechanical stability of integument.13–15 These proteins can be intracellular, transmembrane, or extracellular, and they are used in the formation of the cytoskeleton, cell-cell binding, and cell-matrix interactions. EB follows an autosomal mode of transmission that can be dominant or recessive. The large number of proteins that form part of the adhesive structures of the skin explain the clinical heterogeneity of the disease. More than 1000 mutations in at least 19 genes have been associated with the pathogenesis of EB (Table 1). Recently, a new gene, KLHL24, has been linked to skin fragility.13 This gene encodes kelch-like protein 24 and causes a new type of basal EBS that is not included in the current classification and so is not mentioned in the following sections.
Recommendations for Diagnosis of Epidermolysis Bullosa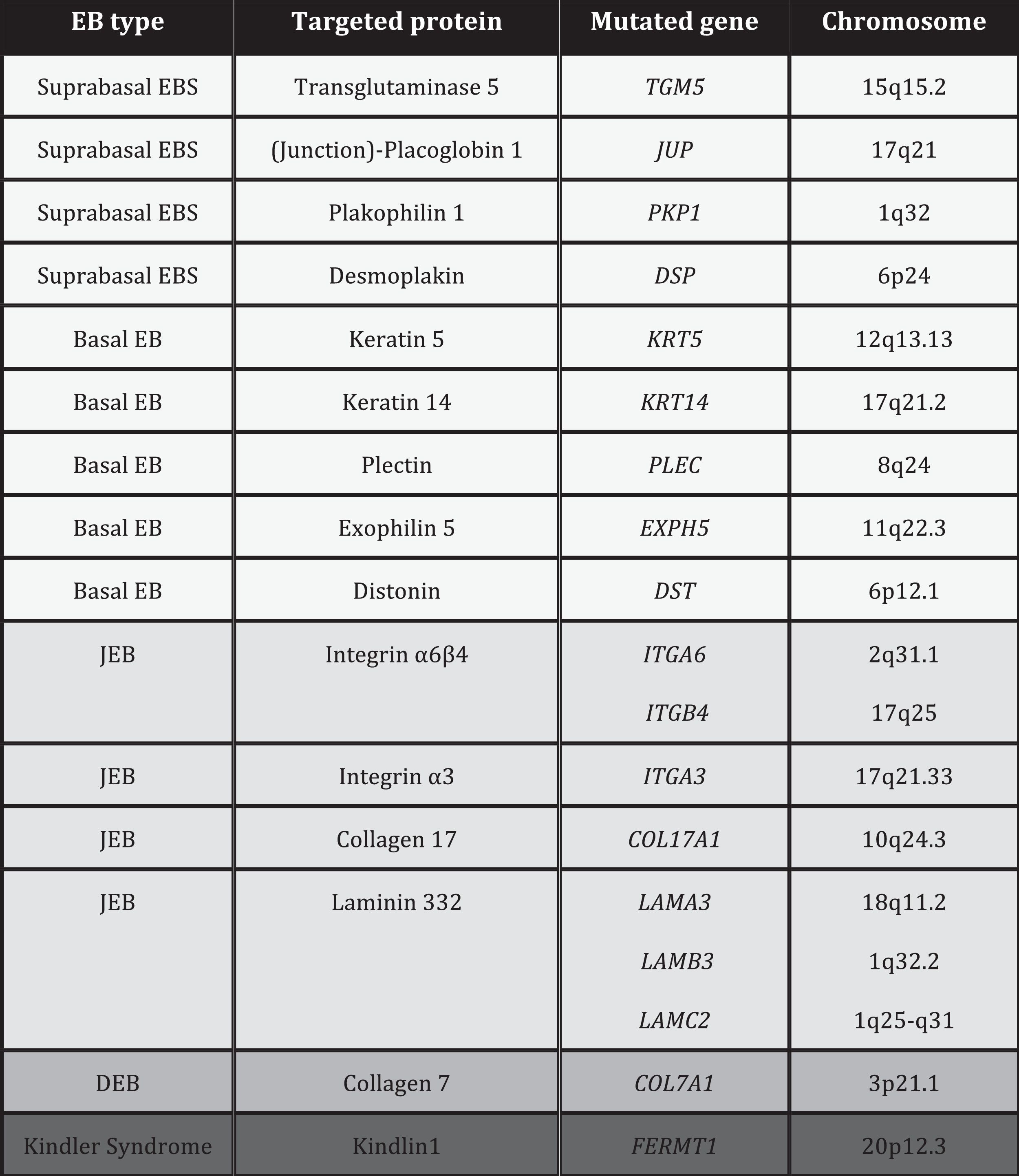
Given the large number of proteins involved in EB, diagnosis is complex and classification has been subject, over the last 20 years, to regular international consensus updates, the most recent being in 2013.2 The most recent classification of EB is based on a layer system (onion skinning) which takes into account sequentially the level of skin cleavage, phenotypic characteristics, mode of inheritance, antigen mapping findings, the gene involved, and the causal mutation of the disease.2 According to these criteria, up to 30 subtypes of EB have been reported.
Faced with the aforementioned complexity, diagnosis of EB requires, in addition to blood sampling for genomic DNA, skin biopsy that includes an area of cleavage between the dermis and epidermis induced by friction at the time the biopsy is taken. This biopsy is used for antigen mapping to establish the cleavage level and to guide diagnosis towards one of the 4 main types of EB and, therefore, delimit the possible candidate genes. Antigen mapping is usually performed by using anti-collagen IV (Col IV) antibodies and a battery of different antigens present in basement membrane proteins implicated in the most common forms of EB. Expression is analyzed by immunofluorescence. In many recessive forms of EB, mutations associated with functional loss generally lead to a reduction in or absence of immunolabeling of a particular protein, that allows to identify the best candidate gene. However, in the dominant forms (and some recessive ones), in general, the results of antigen mapping do not show clear differences in the level of expression of the different proteins analyzed. In these cases, it is essential that the sample includes a recent blister to enable determination of the level of cleavage. Although electron transmission microscopy was initially used for diagnosis, this technique has now been almost completely replaced by antigen mapping.
Finally, identification of the mutation in the gene that encodes the protein whose abnormality is responsible for the disease enables definitive diagnosis, helps (within certain limits) to provide a prognosis and is key for inclusion of patients in clinical trials. There are standard protocols that include polymerase chain reaction and subsequent Sanger sequencing. To analyze the 18 genes implicated in the different subtypes of EB, more than 400 primer pairs are required. This explains the utility of limiting the number of candidate genes through antigen mapping of skin biopsies. The search for mutations is very laborious as more than 1000 have been described. In many cases, new mutations that are associated only with a single family continue to be discovered. Given the limitations of direct sequencing, pathogenic mutations are not identified in approximately 15% of cases. The use of massive next generation sequencing (NGS) currently allows greater sensitivity in the diagnosis of EB.16
In many cases in which the mutation identified is unique to each family (that is, it has not been identified in another patient), it is necessary to rule out a nonpathogenic polymorphic variant. To do this, the presence and frequency of the mutation in the general population of the same origin should be investigated. Furthermore, it is helpful to determine the pathogenic effect of the mutation at the RNA messenger or protein level. For these studies, a second skin biopsy is used to isolate cells from the dermis and epidermis of the patient. It is recommended to obtain this biopsy in the same surgical procedure as the one for immune mapping. The data collected will be of interest for establishing the strategy for laboratory study with the aim of providing appropriate advice and genetic counselling for the patient and their family in each particular situation. Only when the patient and their family have been informed final diagnosis is actually reached.
Table 2 shows the recommended steps for diagnosis of EB.
Recommended Steps in case of EB's suspicion.
| I. Clinical Diagnosis |
| II. Molecular Diagnosis |
| Antigen mapping: selection of the candidate gene |
| Genetic diagnosis: detection of mutations by PCR and sequencing |
| III. Advice and Genetic Counselling |
| The following medical interventions may be required: |
| IV. Prenatal Diagnosis (Invasive and Noninvasive) |
| V. Preimplantation Diagnosis |
Diagnosis of EB begins with a complete clinical history of the patient, including cutaneous and extracutaneous manifestations of the disease, the moment of onset of symptoms, and relevant family history. The family tree should show, among others, the age of the parent, possible consanguinity, and geographical origin of the family.
Diagnosis of the EB subtype is complicated in neonates, as many of the initial symptoms of the disease are common to the 4 main types and it is almost impossible to differentiate between them. It is also essential to perform a differential diagnosis between the hereditary forms of EB and autoimmune blistering diseases.17,18 In the latter case, lesions may arise due to inflammatory action of antibodies that react against certain molecules in the dermal-epidermal junction and not to mutations in the gene that encodes these proteins, as occurs in the hereditary forms. The presence of relevant family medical history is, without doubt, very valuable information.
In adult patients in whom the symptomatology of the disease is completely established, manifestations may be very specific and can guide diagnosis towards one of the subtypes. In the most recent classification,2 each clinical feature in each EB subtype is classified according to severity, time of onset, etc. This information is very useful for differentiating between some subtypes with common features.2 However, in the mild forms of EB, the subtypes can often also be clinically indistinguishable in adults.
Below, we present a summary of the main characteristics of each type and subtype of EB, some illustrative clinical images, and a table (Table 3) to consult the affected proteins in each subtype.
Clinical Classification of EB.
| Type | Subtype | Clinical Subtype | Protein |
|---|---|---|---|
| EBS | |||
| Suprabasal | |||
| Acral peeling skin syndrome (APSS) | Transglutaminase 5 | ||
| EBS superficialis (EBSS) | Unknown | ||
| Acantholytic EBS (EBS-acanth) | Desmoplakin, plakoglobin | ||
| Skin fragility syndromes: | |||
| - Desmoplakin deficiency: skin fragility-woolly hair syndrome | Desmoplakin | ||
| - Plakoglobin deficiency | Plakoglobin | ||
| - Plakophilin deficiency: skin fragility-ectodermal dysplasia syndrome | Plakophilin 1 | ||
| Basal | |||
| Localized EBS (EBS-Loc) | Keratin 5 (K5), keratin 14 (K14) | ||
| Generalized severe EBS (EBS-gen sev) | Keratin 5 (K5), keratin 14 (K14) | ||
| Generalized intermediate EBS (EBS-gen intermed) | Keratin 5 (K5), keratin 14 (K14) | ||
| EBS with mottled pigmentation (EBS-MP) | Keratin 5 (K5) | ||
| Migratory circinate EBS (EBS-migr) | Keratin 5 (K5) | ||
| Autosomal recessive K14 EBS (EBS-AR K14) | Keratin 14 (K14) | ||
| EBS with muscular dystrophy (EBS-MD) | Plectin 1 | ||
| EBS with pyloric atresia (EBS-PA) | Plectin, α6β4 integrin | ||
| Ogna EBS (EBS-Og) | Plectin | ||
| Autosomal recessive-BP230 deficiency EBS (EBS-AR BP230) | Bullous pemphigoid antigen-1 (BP230) | ||
| Autosomal-recessive exophilin 5 deficiency EBS (EBS-AR exophilin 5) | Exophilin 5 | ||
| JEB | |||
| Generalized | |||
| Generalized severe JEB (JEB-sev gen) | Laminin 332 | ||
| Generalized intermediate JEB (JEB-gen intermed) | Laminin 332, collagen XVII | ||
| JEB with pyloric atresia (JEB-PA) | α6β4 integrin | ||
| Localized | |||
| Localized JEB (JEB-loc) | Laminin 332, collagen XVII, β4 integrin | ||
| JEB inversa (JEB-inv) | Laminin 332 | ||
| LOC syndrome | Laminin 332, α3 chain | ||
| DEB | |||
| Dominant | |||
| Generalized DDEB (DDEB-gen) | Collagen VII | ||
| Localized DDEB (DDEB-loc) | Collagen VII | ||
| Acral DDEB (DDEB-ac) | Collagen VII | ||
| Pretibial DDEB (DDEB-pt) | Collagen VII | ||
| DDEB pruriginosa (DDEB-pr) | Collagen VII | ||
| Nails only DDEB (DDEB-na) | Collagen VII | ||
| Newborn (DDEB-BDN) | Collagen VII | ||
| Recessive | |||
| Generalized severe RDEB (RDEB-gen sev) | Collagen VII | ||
| Generalized intermediate RDEB (RDEB-gen intermed.) | Collagen VII | ||
| RDEB inversa (RDEB-inv) | Collagen VII | ||
| Localized RDEB (RDEB-loc) | Collagen VII | ||
| Pretibial RDEB (RDEB-pt) | Collagen VII | ||
| RDEB pruriginosa (RDEB-pr) | Collagen VII | ||
| RDEB centripetalis (RDEB-ce) | Collagen VII | ||
| Newborn (RDEB-BDN) | Collagen VII | ||
| KS | |||
| Kindlin 1 |
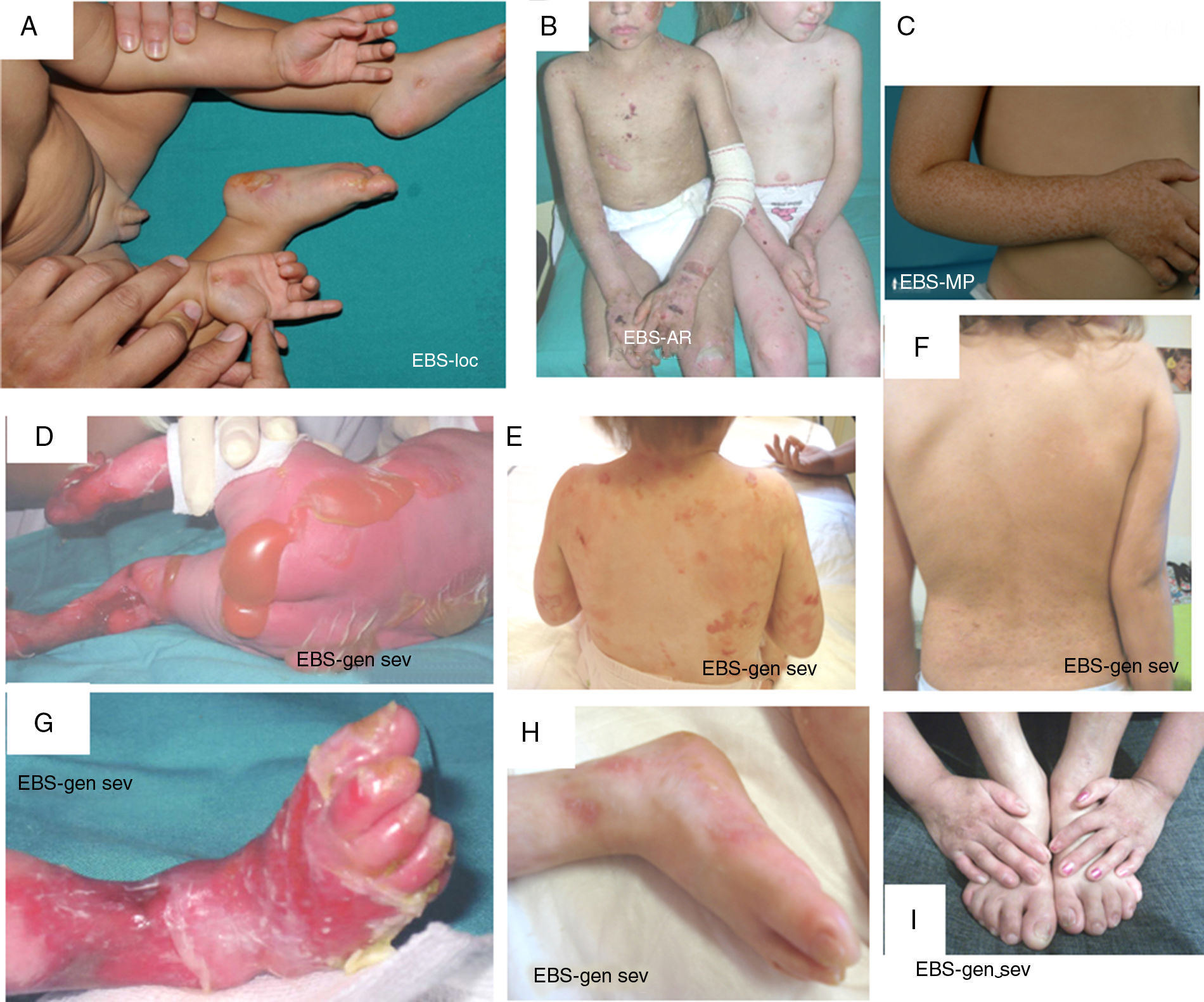
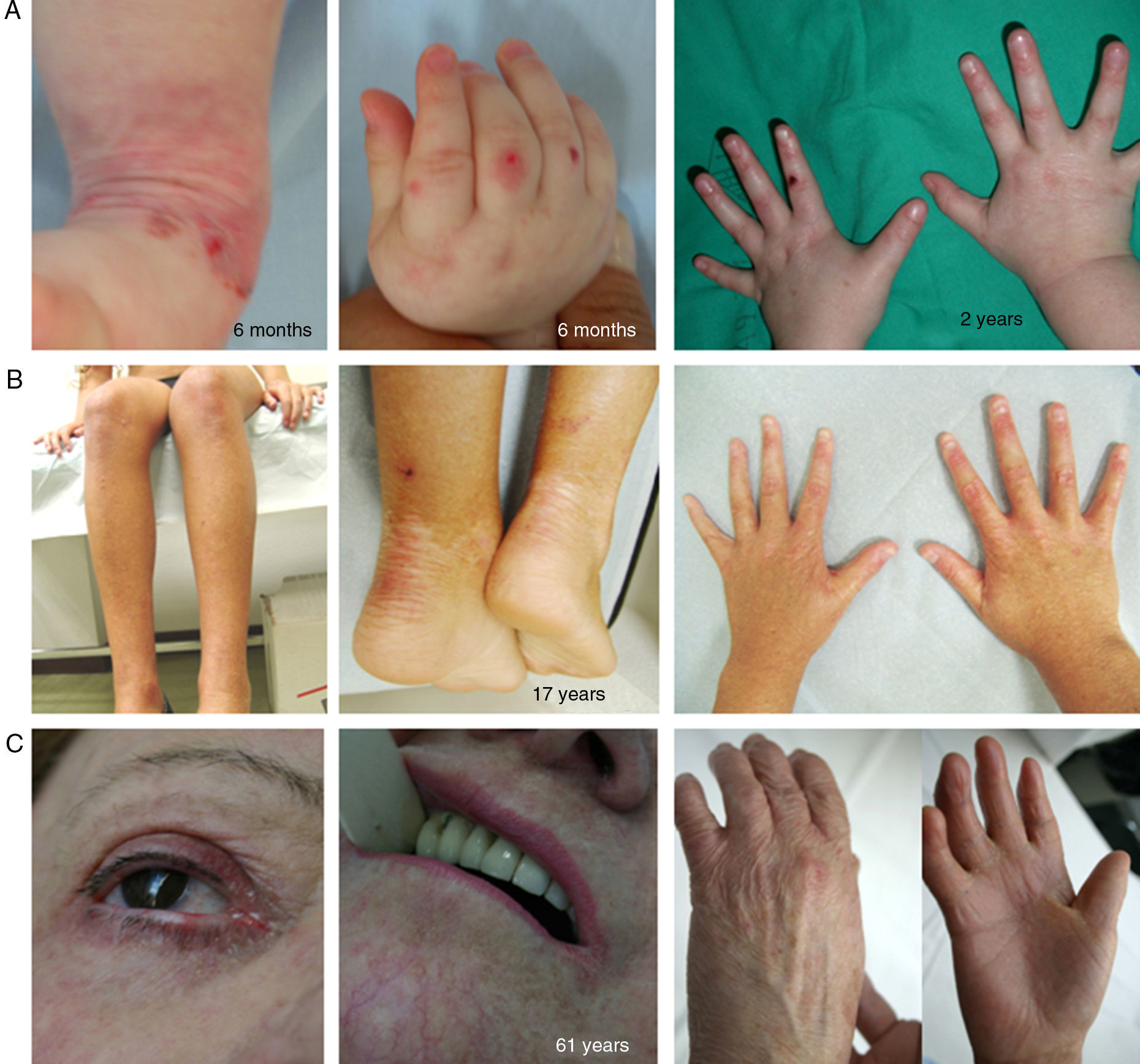
The term EBS includes all forms of EB in which cutaneous fragility is confined to the epidermis. In the vast majority of EBS forms, cleavage is detected histologically at the basal level and the disease follows a pattern of autosomal dominant transmission. Autosomal recessive transmission in EBS accounts for only 5% of cases. Although onset of clinical manifestations occurs, in general, at birth or during the neonatal period (Fig. 2), the first manifestations may appear in adults. In general, blistering in EBS is induced by trauma and, may rarely occur spontaneously. Scarring, milia, and dystrophic nails are usually less frequent in EBS than in JEB and DEB.
 are causative of the disease in all patients.19,20 Photographs obtained with signed informed consent (Source: u714-CIBERER-CIEMAT-UC3M-IISFJD). A, Palmoplantar involvement of a patient with localized EBS during infancy. B, Generalized involvement of 2 siblings with EBS, of consanguineous parents, with autosomal recessive transmission19. C, Mottled pigmentation in a patient with EBS-MP.21 D-G, Extensive involvement in a neonate with generalized severe EBS. E-F-H-I, Progression during infancy. E, Widely distributed herpetiform lesions on the patient")
Clinical manifestations of basal EBS. The keratin-encoding genes (KRT5, KRT14) are causative of the disease in all patients.19,20 Photographs obtained with signed informed consent (Source: u714-CIBERER-CIEMAT-UC3M-IISFJD).
A, Palmoplantar involvement of a patient with localized EBS during infancy. B, Generalized involvement of 2 siblings with EBS, of consanguineous parents, with autosomal recessive transmission19. C, Mottled pigmentation in a patient with EBS-MP.21 D-G, Extensive involvement in a neonate with generalized severe EBS. E-F-H-I, Progression during infancy. E, Widely distributed herpetiform lesions on the patient's back.
Localized EBS is the most common form and it is characterized by blisters confined to the palms and soles and, in some cases, by oral erosions or blisters during infancy. Lesions can be present in any part of the body as a result of skin trauma. Severe generalized EBS, a less frequent subtype, is usually associated with marked morbidity and mortality during the neonatal period or early infancy. Its most distinctive feature is the formation of intact blisters grouped in an arch-shaped distribution (herpetiform). Patients with this form usually develop skin thickening on the palms and soles, giving rise to keratosis that may resolve on reaching adulthood. Some patients have nail distrophy atrophic scarring, milia, mucosal involvement (laryngeal stenosis), anemia, and retarded growth. Unlike the severe form, generalized intermediate EBS presents with nonherpetiform blisters, and anemia and growth retardation are uncommon.2
Other less frequent basal EBS subtypes are EBS with mottled pigmentation, migratory circinate EBS, autosomal recessive EBS (EBS-AR), EBS with muscular dystrophy, EBS with pyloric atresia, EBS with BP230 deficiency, and EBS with exophilin 5 deficiency.2 Recently, a new protein (kelch-like protein 24) has been identified, associated with a basal EBS subtype with distinctive clinical features13 compared with the classic forms of EBS (alopecia, follicular and cutaneous atrophy).
Currently, the term EBS also encompasses those forms in which mechanical fragility, unlike those described previously, occurs at the suprabasal level. This suprabasal EBS subtype includes the acral form, EBS superficialis, the acantholytic form, and skin fragility syndromes arising from desmoplakin, plakoglobin, and plakophilin deficiencies4 respectively (see Tables 1 and 3).
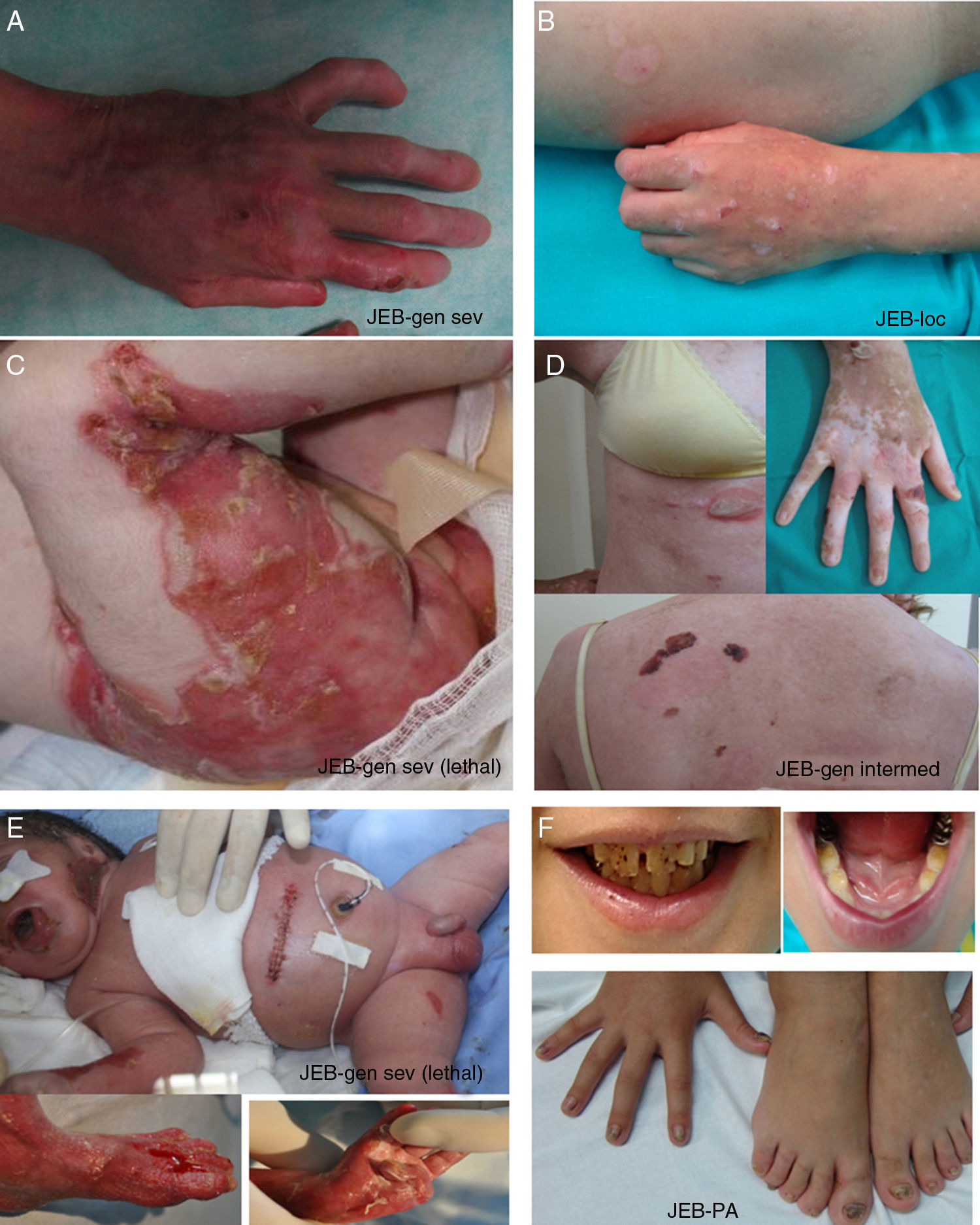
Junctional Epidermolysis BullosaThe most prominent clinical feature in all subtypes of JEB is enamel hypoplasia, a useful distinguishing characteristic for diagnosis of this type of EB (Fig. 3F). JEB can also be subdivided into several subtypes, all of which are of autosomal recessive transmission.
. A-B, Severe generalized JEB caused by mutations in the LAMC2 gene with absence of Lam 332, specifically the γ2 chain, which led to neonatal death in one of the patients. C, Intermediate generalized JEB. D, Localized JEB. In both patients, mutations were found in the COL17A1 gene, leading to reduced expression and complete absence of Col17, respectively. Note the postinflammatory hypopigmentation. E-F, JEB with pyloric atresia caused by mutations in the ITGB4 gene; protein expression was comparable to that of control in patient F and absent in the lethal form (not shown).22 Both patients underwent corrective surgery for pyloric atresia.")
Clinical manifestations of JEB. Photographs obtained with signed informed consent (Source: U714-CIBERER-CIEMAT-UC3M-IISFJD). A-B, Severe generalized JEB caused by mutations in the LAMC2 gene with absence of Lam 332, specifically the γ2 chain, which led to neonatal death in one of the patients. C, Intermediate generalized JEB. D, Localized JEB. In both patients, mutations were found in the COL17A1 gene, leading to reduced expression and complete absence of Col17, respectively. Note the postinflammatory hypopigmentation. E-F, JEB with pyloric atresia caused by mutations in the ITGB4 gene; protein expression was comparable to that of control in patient F and absent in the lethal form (not shown).22 Both patients underwent corrective surgery for pyloric atresia.
The most severe subtype is the severe generalized which is present from birth and can affect the entire skin (Fig. 3A-B). A very significant finding in this subtype is the appearance of abundant symmetrically distributed granulation tissue in the mouth, central region of the face and nose, the upper part of the back, axillas, and inguinal folds. The clinical picture includes oral blisters, microstomia, and ankyloglossia, although these latter manifestations are not as marked as in the recessive forms of DEB. Extracutaneous manifestations can also affect other organs such as the esophagus, eyes, upper respiratory tract, and genitals. Patients may also present with growth retardation and multifactorial anemia. There is a high risk of death during infancy (up to 2 years of age) as a result of growth retardation, infections, pneumonia, and airway obstruction.
Generalized intermediate JEB, evident from birth, is the most common of the junctional variants. This generalized disorder presents as blisters, atrophic scars, and dystrophic or absent nails (Fig. 3D). The patients usually also present with postinflammatory hypopigmentation or depigmentation and, in some cases, scarring alopecia of the scalp. The risk of death due to airway occlusion or other complications is much lower than in the generalized form, as is the risk of anemia and growth retardation. Some patients, however, may develop squamous cell carcinoma.
Another clinically important subtype is JEB inversa, which presents as severe blisters confined to intertriginous sites of the skin, esophagus, and vagina. JEB-LOC syndrome is characterized by the formation of localized blisters on the face and neck with granulation tissue, upper airway abnormalities, nail abnormalities, and tooth enamel hypoplasia.2 JEB with pyloric atresia presents with generalized blisters from birth and congenital atresia of pylorus sphincter. Congenital abnormalities of the genitourinary tract may also be present with variable prognosis from a milder phenotype to neonatal death (Fig. 3E-F). It is worth mentioning that there is a form of EB associated with pyloric atresia in which skin cleavage occurs in the basal epidermal layer and not in the dermal-epidermal junction. This form is therefore considered a subtype of EBS (EBS with pyloric atresia; Tables 1 and 3).
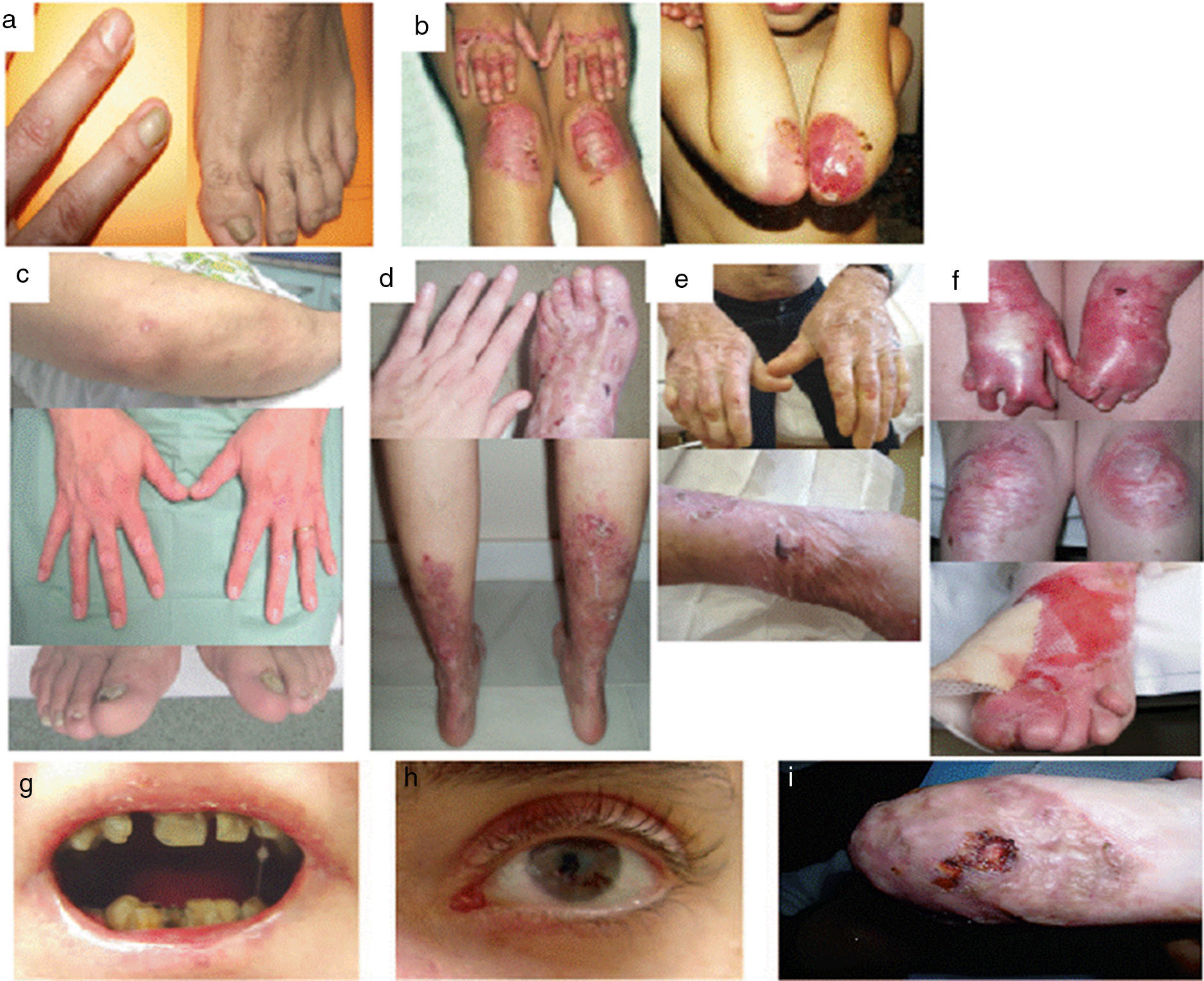
Dystrophic Epidermolysis BullosaDEB is divided into 2 large groups, dominant and recessive, according to the pattern of genetic of transmission (Table 3, Fig. 4).1,2
. Mutations in the gene that encodes collagen type VII (COL7A1) is the cause of the disease in all patients.23,24 A, Dominant DEB, nails only. B, Dominant DEB, generalized. C, Recessive DEB, localized. D, Recessive DEB, pretibial. E, Recessive DEB, generalized (non Hallopeau-Siemens syndrome). F, Recessive DEB, generalized severe (Hallopeau-Siemens). F, Recessive DEB, generalized severe (Hallopeau-Siemens). G-I, Other complications in patients with severe generalized RDEB. G, Microstomia with dental abnormalities and caries. H, Scarring and ocular blisters (lower eyelid). I, Squamous cell carcinoma on the stump of the left hand.")
Clinical Manifestations of DEB. Clinical photographs of Spanish patients obtained with signed informed consent (Source: U714-CIBERER-CIEMAT-UC3M-IISFJD). Mutations in the gene that encodes collagen type VII (COL7A1) is the cause of the disease in all patients.23,24 A, Dominant DEB, nails only. B, Dominant DEB, generalized. C, Recessive DEB, localized. D, Recessive DEB, pretibial. E, Recessive DEB, generalized (non Hallopeau-Siemens syndrome). F, Recessive DEB, generalized severe (Hallopeau-Siemens). F, Recessive DEB, generalized severe (Hallopeau-Siemens). G-I, Other complications in patients with severe generalized RDEB. G, Microstomia with dental abnormalities and caries. H, Scarring and ocular blisters (lower eyelid). I, Squamous cell carcinoma on the stump of the left hand.
The most severe subtype of dominant DEB (DDEB) is the generalized, which is characterized by the presence of blisters distributed throughout the body from birth. Milia atrophy, scarring, and dystrophic nails are also associated at a later age (Fig. 4B). Recurrent blisters and erosions in the esophagus is common. Other less severe types of DDEB are the nails only form (Fig. 4A), the acral form, localized DDEB with blisters confined to the hands and feet (Fig. 4C), the pretibial form, with minor plaque-like lesions, often of violaceous appearance (Fig. 4D), pruriginous form, which gives rise to untreatable pruritus, and the newborn form with generalized blisters accompanied by focal scarring which usually disappears after between 6 and 24 months of life.
Recessive DEB (RDEB), also divided into several subgroups, can show a wide range of severity, from simple nail dystrophy to generalized blistering and hand and feet deformities (pseudosyndactyly). The disease can also affect the mucosa and, in the most severe forms, is associated with a high risk of developing squamous cell carcinoma. The most frequent subtype is the generalized severe which is evident from birth, with blisters that progressively spread throughout the body and result in atrophic scarring. This subtype presents with granulation tissue, above all in chronic wounds, as well as joint contractures and development of pseudosyndactyly (Fig. 4F). Patients usually develop extensive caries, severe ankyloglossia, and microstomia (Fig. 4G) associated with deficient food intake. Other extracutaneous manifestations are corneal blisters, esophageal stenosis, chronic renal failure, multifactorial anemia, and retarded growth. This subtype, which is the most severe form of RDEB, is often complicated by the development of metastatic squamous cell carcinomas (Fig. 4I) and premature death may occur. The generalized intermediate subtype is less severe (Fig. 4E), and generally does not present with retarded growth or anemia, and the frequency of esophageal stenosis as well as skin involvement is lower. However, there is still a significant risk of developing squamous cell carcinoma. The less common subtypes are RDEB centripetalis, which starts in the acral areas and extends to the trunk, and RDEB inversa, in which the appearance of blisters is limited to intertriginous areas, the base of the neck, the top of the back, and the lumbosacral area. Patients with RDEB inversa are prone to develop blisters in the oral cavity, esophagus, and genitourinary tract, and may end up developing debilitating stenosis of the esophagus and vagina.
Kindler SyndromeKS is a recessive autosomal genodermatosis that is included in the EB classification given its clinical and biological features. This means that patients with this syndrome (fewer than 200 individuals throughout the world) are attended in EB reference centers.1,2 KS appears at birth with the presence of blisters that can have a generalized distribution and often disappear as the patient gets older (Fig. 5). The course of this form is characterized by the progressive development of poikiloderma, photosensitivity, premature skin aging, and keratoderma.25 Patients with KS can present with atrophic scarring, nail dystrophy, contractures, and interdigital synechia. Complications such as severe colitis, esophagitis, urethral stenosis, gingival hyperplasia, and squamous cell carcinomas can be associated with KS.1,2 The pro-oxidative state, described recently in epithelial cells of patients with KS, may explain the pathognomonic features not associated with skin fragility.26,27
. Mutations in the gene that encodes kindlin 1 (KIND1) is the cause of the disease in all patients.26–28 A, Erosions and premature aging of the skin, incipient already in infancy, on the backs of the hands. B, Evident cutaneous aging and poikiloderma along with nail dystrophy in a young patient. C, Ocular involvement, poikiloderma, interdigital synechia, and contracture of the hands in a female patient in her sixties.")
Clinical Manifestations of KS. Clinical photographs of patients obtained with signed informed consent (Source: U714-CIBERER-CIEMAT-UC3M-IISFJD). Mutations in the gene that encodes kindlin 1 (KIND1) is the cause of the disease in all patients.26–28 A, Erosions and premature aging of the skin, incipient already in infancy, on the backs of the hands. B, Evident cutaneous aging and poikiloderma along with nail dystrophy in a young patient. C, Ocular involvement, poikiloderma, interdigital synechia, and contracture of the hands in a female patient in her sixties.
The first step recommended in the diagnosis of EB is a clinical classification of patients with skin fragility and, thus suspicion of possible abnormalities in dermal-epidermal anchoring proteins.14,15 Diagnosis continues with initial classification in the 4 main types of EB through identification of the level at which cleavage occurs by immunofluorescence (Table 4).
Cleavage Level in Each of the Four Main Types of EB.
| Main Types of EB | Level of blister formation |
|---|---|
| Simplex EB | Intraepidermal |
| Junctional EB | Intralamina lucida (basement membrane) |
| Dystrophic EB | Sublamina densa (dermal) |
| Kindler Syndrome | Multiple levels (intraepidemral, intralamina lucida, and sublamina densa) |
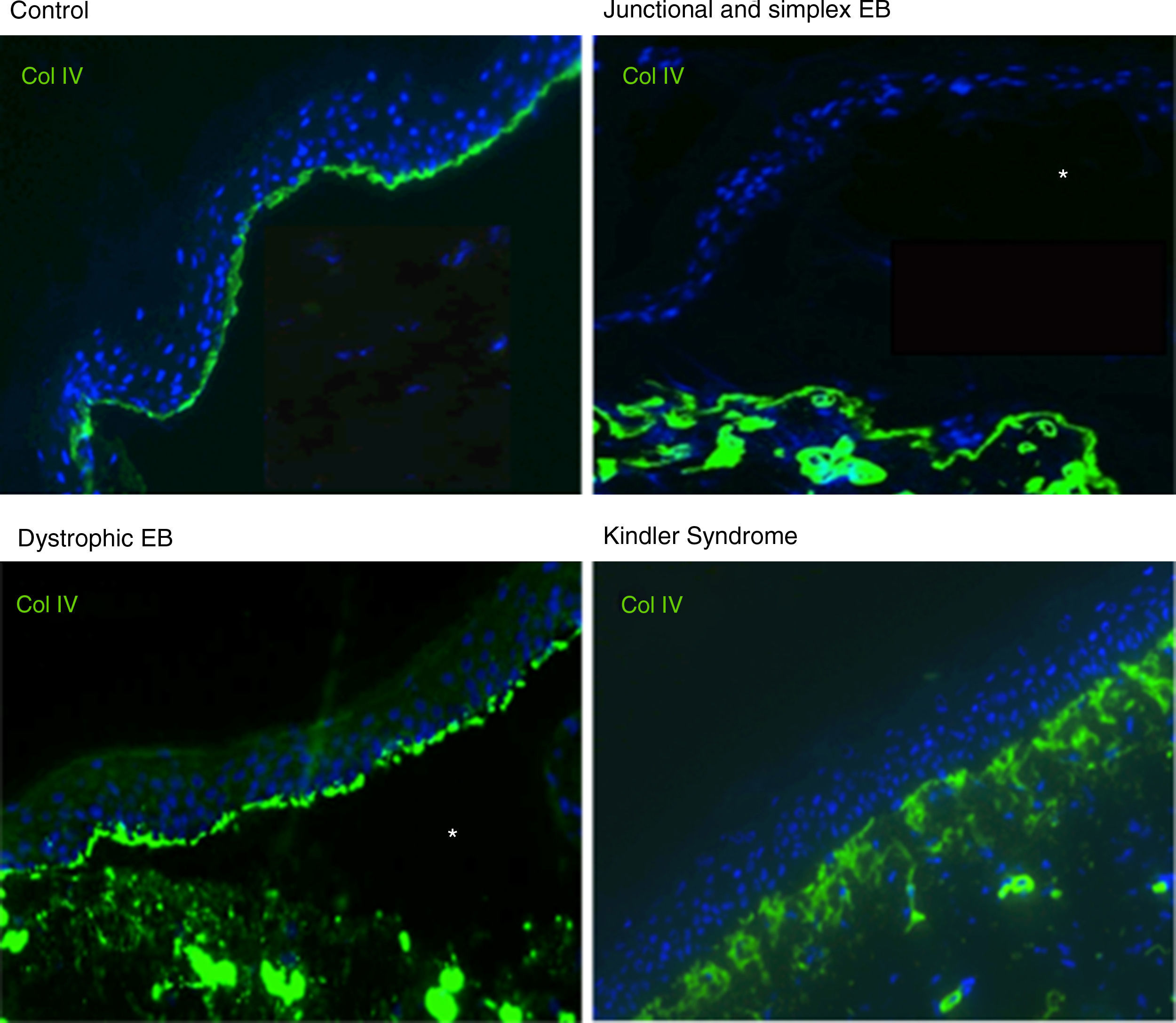
Antigen mapping is performed by using histological sections from biopsies of cryopreserved skin that includes a blister just before sampling. These sections are incubated with a battery of antibodies against Col IV and different protein implicated in EB. The antibody against Col IV, protein present in the lamina densa of the dermal-epidermal junction, enables us to unequivocally identify DEB (labeling located in the roof of the blister) and KS (reduplication of the basement membrane). The use of this antibody does not distinguish between EBS and JEB, as the labeling is present in the floor of the blister in both cases (Fig. 6).
.")
Col IV Immunofluorescence. Col IV can be observed at the floor of the blister in both JEB and EBS. In DEB, Col IV is observed in the roof of the blister, whereas in KS, a reduplication of the basement membrane is observed, distinctive for this disease. Source: Dr. Marta García (U714-CIBERER-CIEMAT-UC3M-IISFJD).
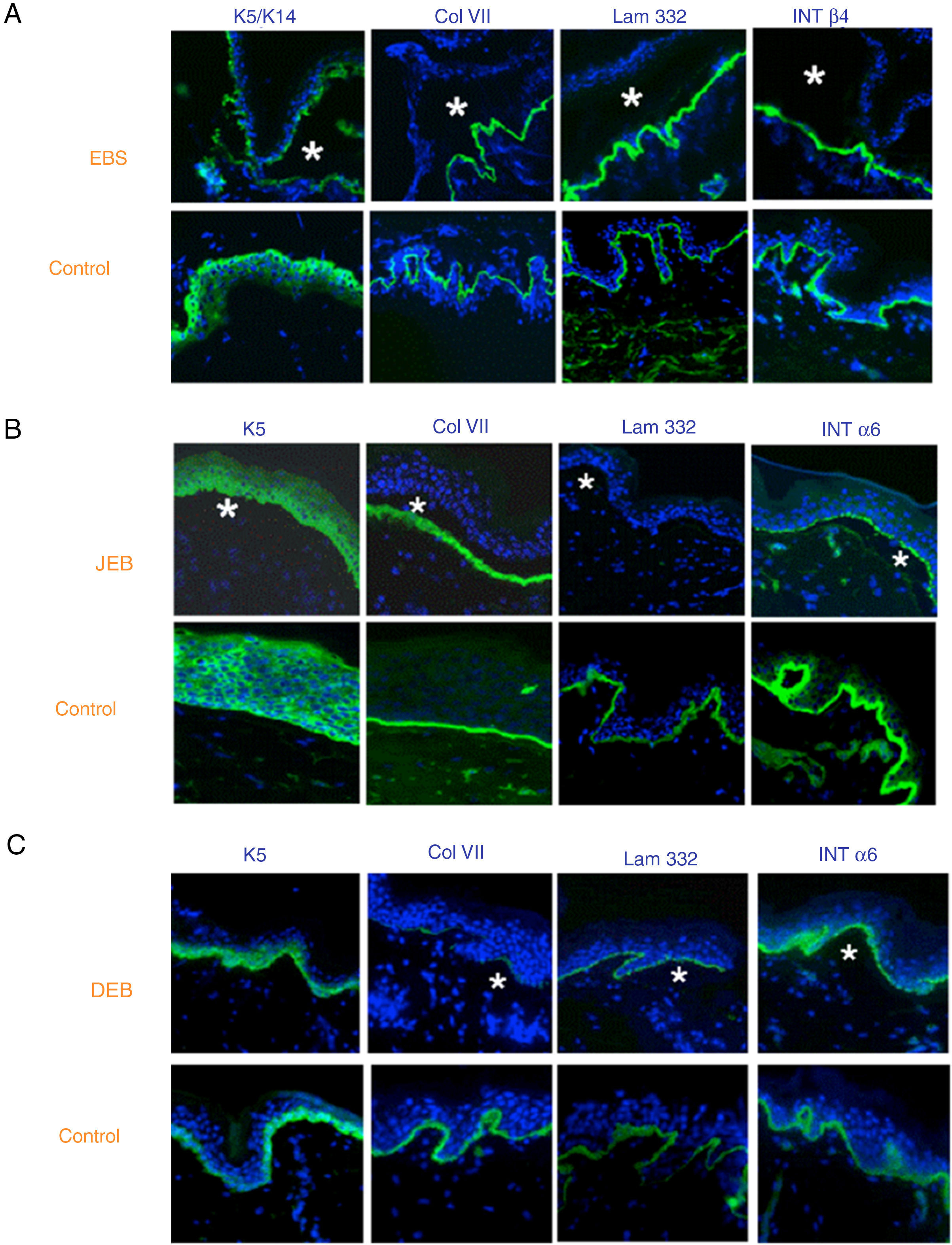
The use of specific antibodies for the proteins implicated in EB and their expression pattern in the roof or floor of the blister helps to differentiate between EBS, JEB, and DEB (Figs. 7 and 8) also to delimit candidate genes.

 in histological sections of biopsies of cryopreserved skin. Microphotographs: Dr. Marta García (U714-CIBERER-CIEMAT-UC3M-IISFJD). A, Immunophenotype of basal EBS. The K5 and K14 keratins mark the floor and roof of the blister as a result of intraepidermal cleavage. Labeling of the remaining proteins is localized in the floor of the blister. The result indicates that the candidate genes in the initial screening are KRT5 and KRT14. B, Immunophenotype of JEB. As a result of cleavage of the intralamina lucida, labeling of the keratins is restricted to the blister roof while that of collagen VII remains in the lister floor. In this case, the absence of labeling of laminin 332 indicates that the candidate genes are LAMA3, LAMB3, and LAMC3, and that the molecular subclassification is JEB (see Table 5). Note that the absence of Lam332 can affect expression of integrins. C, Immunophenotype of DEB. As a result of cleavage at the level of the papillary dermis, all markers map to the roof of the blister. This result indicates that the candidate gene is COL7A1. The almost complete absence of Col VII is consistent with generalized severe DEB (see Table 5).")
Classification of EB and selection of candidate gene by antigen mapping (first screening) in histological sections of biopsies of cryopreserved skin. Microphotographs: Dr. Marta García (U714-CIBERER-CIEMAT-UC3M-IISFJD). A, Immunophenotype of basal EBS. The K5 and K14 keratins mark the floor and roof of the blister as a result of intraepidermal cleavage. Labeling of the remaining proteins is localized in the floor of the blister. The result indicates that the candidate genes in the initial screening are KRT5 and KRT14. B, Immunophenotype of JEB. As a result of cleavage of the intralamina lucida, labeling of the keratins is restricted to the blister roof while that of collagen VII remains in the lister floor. In this case, the absence of labeling of laminin 332 indicates that the candidate genes are LAMA3, LAMB3, and LAMC3, and that the molecular subclassification is JEB (see Table 5). Note that the absence of Lam332 can affect expression of integrins. C, Immunophenotype of DEB. As a result of cleavage at the level of the papillary dermis, all markers map to the roof of the blister. This result indicates that the candidate gene is COL7A1. The almost complete absence of Col VII is consistent with generalized severe DEB (see Table 5).
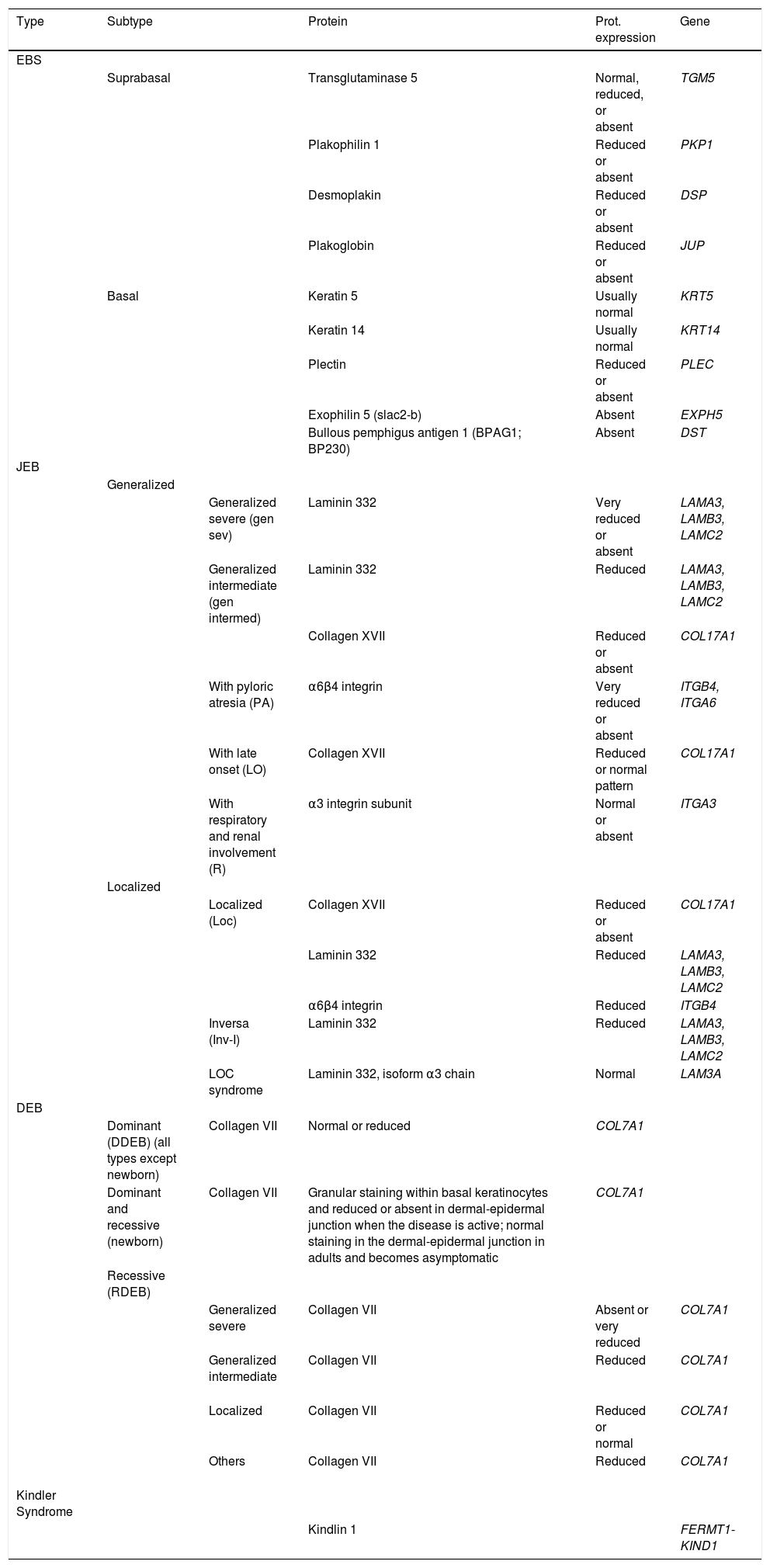
In some cases, the results of immunofluorescence enable to further refine the subclassification, as decreased intensity (Table 5) or complete lack of labeling of a given protein consistently points its encoding gene as the candidate gene. The complete lack of labeling generally corresponds to null mutations. In other cases, complete proteins are produced with an altered amino acid sequence and its expression may be similar to the one in a healthy individual. The process for selection of the candidate gene is described in Figure 7.
Molecular Classification of EB.
| Type | Subtype | Protein | Prot. expression | Gene | |
|---|---|---|---|---|---|
| EBS | |||||
| Suprabasal | Transglutaminase 5 | Normal, reduced, or absent | TGM5 | ||
| Plakophilin 1 | Reduced or absent | PKP1 | |||
| Desmoplakin | Reduced or absent | DSP | |||
| Plakoglobin | Reduced or absent | JUP | |||
| Basal | Keratin 5 | Usually normal | KRT5 | ||
| Keratin 14 | Usually normal | KRT14 | |||
| Plectin | Reduced or absent | PLEC | |||
| Exophilin 5 (slac2-b) | Absent | EXPH5 | |||
| Bullous pemphigus antigen 1 (BPAG1; BP230) | Absent | DST | |||
| JEB | |||||
| Generalized | |||||
| Generalized severe (gen sev) | Laminin 332 | Very reduced or absent | LAMA3, LAMB3, LAMC2 | ||
| Generalized intermediate (gen intermed) | Laminin 332 | Reduced | LAMA3, LAMB3, LAMC2 | ||
| Collagen XVII | Reduced or absent | COL17A1 | |||
| With pyloric atresia (PA) | α6β4 integrin | Very reduced or absent | ITGB4, ITGA6 | ||
| With late onset (LO) | Collagen XVII | Reduced or normal pattern | COL17A1 | ||
| With respiratory and renal involvement (R) | α3 integrin subunit | Normal or absent | ITGA3 | ||
| Localized | |||||
| Localized (Loc) | Collagen XVII | Reduced or absent | COL17A1 | ||
| Laminin 332 | Reduced | LAMA3, LAMB3, LAMC2 | |||
| α6β4 integrin | Reduced | ITGB4 | |||
| Inversa (Inv-I) | Laminin 332 | Reduced | LAMA3, LAMB3, LAMC2 | ||
| LOC syndrome | Laminin 332, isoform α3 chain | Normal | LAM3A | ||
| DEB | |||||
| Dominant (DDEB) (all types except newborn) | Collagen VII | Normal or reduced | COL7A1 | ||
| Dominant and recessive (newborn) | Collagen VII | Granular staining within basal keratinocytes and reduced or absent in dermal-epidermal junction when the disease is active; normal staining in the dermal-epidermal junction in adults and becomes asymptomatic | COL7A1 | ||
| Recessive (RDEB) | |||||
| Generalized severe | Collagen VII | Absent or very reduced | COL7A1 | ||
| Generalized intermediate | Collagen VII | Reduced | COL7A1 | ||
| Localized | Collagen VII | Reduced or normal | COL7A1 | ||
| Others | Collagen VII | Reduced | COL7A1 | ||
| Kindler Syndrome | |||||
| Kindlin 1 | FERMT1-KIND1 |
Although immunophenotyping is currently not essential thanks to massive sequencing, it is still recommended as first step prior to genetic study. Immunophenotyping helps identify the implicated protein and, therefore, the candidate gene.23,29,30Table 5 shows the different EB subtypes according to molecular criteria: affected protein, level of expression in the skin, and encoding genes.
Genetic DiagnosisAfter determining the affected protein in the patient, we can proceed to genetic analysis through sequencing of the candidate gene. The search begins with focus on the most common mutations and ends with complete sequencing of the gene, if necessary. Currently, the search for the mutation is performed by Sanger sequencing, although new techniques are already being incorporated, such as NGS, which can include the search for mutations on different genes at the same time.16 NGS is a very useful step forwards, as molecular study can be performed directly, regardless of the diagnosis of the immunophenotyping analysis of the skin biopsy. NGS techniques, through panels that include at least all the coding sequences of known EB-genes, enable diagnosis of a larger number of cases in what is a heterogeneous disease. Even so, and although this approach can increase the proportion of patients diagnosed with genetic diseases, NGS will not be able to identify the mutation in all cases, as occurs in some other genetic diseases. To find a solution in cases in which the pathogenic mutation is not identified through direct gene sequencing, complementary techniques are used for the search for large deletions and duplications, such as multiplex ligation probe amplification or microarray-based comparative genomic hybridization. The incorporation of the mutation into the final classification of EB is critical for providing a rigorous genetic counseling, as it determines the mode of genetic transmission and also enables a theoretical prognosis for the progression of the disease. Prognosis made according to genetic findings should be treated with caution as environmental factors and other genetic modifiers may play a key role in disease progression.2
As mentioned repeatedly, there are many genes implicated in EB and, furthermore, there are many possible mutations in each one of these15; therefore, EB shows heterogeneity in terms of genetic alleles (many possible mutations) and of loci (different genes). All types of mutation have been implicated in EB, from premature stop codons to changes in reading frames, among others. The location is also diverse and may affect both the coding sequence and promotor regions, and even intronic regions. For proper assessment and interpretation of the pathogenic potential of a mutation present in a patient, general genetic databases such as the HGMD (HGMD® Human Gene Mutation Database Professional; http://www.biobase-international.com/product/hgmd) should be searched. These databases include all mutations published as causative of genetic diseases in humans. There are also other more specific databases such as the database of COL7A1 mutations associated with the International Registry of DEB Patients and COL7A1 Mutations31 (http://www.deb-central.org/molgenis.do) or the Human Intermediate Filaments Database32 (http://www.interfil.org). In the event that a mutation has not been reported in the general or specific databases, nor has it been included as a polymorphism in the Short Genetic Variation database (DbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/Variations_Survey.html), the presence should be ruled out in a minimum of 100 control individuals from a population with the same genetic origin. Although not always possible, it is recommended to confirm the pathogenic effect of the mutation at the RNA messenger or protein level.
Starting from the premise that there are more than 1000 mutations described in at least 19 genes, it is very important to have all the information available to optimize the search and perform the risk calculations. The number of new mutations identified increases year after year: in the case of some genes, for example the COL7A1 gene, these mutations are mostly specific to each family. Other information to bear in mind is the frequency of mutations described according to a geographic location or according to a given ethnic origin. For instance in the COL7A1 gene, was observed that the pathogenic c.6527insC mutation is present in 46.3% of alleles of Spanish patients with RDEB being more prevalent in the south-west of Spain (Extremadura and Andalusia). This very frequent mutation, originates from a single common ancestor is currently being investigated in the initial screening for the study of Spanish patients with DEB.23,33–35 This same mutation has also been found with a high recurrence in Chile,36 possibly as a result of waves of Spanish migration to Latin America in different periods. There are other frequent mutations in Spain and, although their prevalence is much lower, screening for them is also included in the study of possible carriers of a COL7A1 mutation. Absence of these recurrent mutations does not rule out the possibility that the proband is a carrier of any other mutation in the COL7A1 gene, but it does drastically reduce the individual risk.
In Spain, there were no reference centers for EB47 until this year, when the Hospital Universitario de la Paz in Madrid and the Hospital Clínic i Provincial de Barcelona and Hospital de Sant Joan de Déu in Catalonia were designated as such. The research group of Prof. Marcela del Río (termeg.uc3m.es) has, however, been performing genetic diagnosis of EB since 2006 (ORPHA361523, ORPHA361536, ORPHA362075, and ORPHA361538), is implicated in research projects aiming to develop treatments for RDEB (TRA-049, CELLCAM, ORPHA300805, GENEGRAFT; ORPHA437567; ORPHA437569) and the national EB registry (https://registroraras.isciii.es). This group, part of the CIBER on Rare Disease Network (www.ciberer.es), is a partner of the international EB CLINET-Clinical Network of EB Centres and Experts (http://www.eb-haus.org/en/eb-clinet.html;http://www.eb-clinet.org/maps/eb-clinical-contacts.html). The group conducts the genetic studies in collaboration with dermatology and genetics departments of several hospitals throughout Spain, and in particular with the Pediatric Dermatology Division of the Hospital La Paz in Madrid (Dr. Raúl de Lucas), recently designated as reference center and with the Genetics Department of the Fundación Jiménez Dáiz (Dr. María José Trujillo-Tiebas; Dr. Carmen Ayuso), in coordination with the Spanish EB patient association, DEBRA España (www.pieldemariposa.es; info@debra.es). This cooperative activity at national and international level has enabled the group to develop a profound knowledge of the disease.8,19,21–26,33,35,37–51 Thus, when EB is suspected, in addition to requesting information from DEBRA-España, it may be helpful to contact Dr. María José Escámez (mj.escamez@ciemat.es), the coordinator of molecular studies conducted by the group, to ensure the most appropriate approach to the genetic study.
Advice and Genetic CounsellingOnce the causative mutation has been identified, it is essential to verify the hereditary pattern to establish the risk of recurrence in future siblings or offspring.
Autosomal Dominant Epidermolysis BullosaTo date dominant forms for EBS and DEB has been described. The dominant forms are the most common ones in EBS whereas they account for a limited number of cases of DEB, for example between 7% and 8% in the Spanish cohort (126 families with DEB have been diagnosed).23 As in any genetic disease with autosomal dominant transmission, a single mutation in one of the copies of the gene is sufficient to cause the disease. In these cases, the affected transmit the mutation (and the disease) to their offsprings with a probability of 50% in each pregnancy. Generally, in DDEB the affected individuals have inherited the disease from one of their progenitors, who is also affected (dominant mutation inherited). However, in dominant EBS, in many cases, the patient is the first case in the family due to a de novo mutation in the germline of unaffected parents (10 families with EBS with de novo KRT5 and KRT10 mutations out of a total of 23 families with mutations in these genes). This is because the genes that encode keratins K5 and K14 have sites well known for genetic instability (hotspots) that are more susceptible to mutations in the gametes of the parents. This phenomenon also occurs with the gene that encodes Col VII, although less frequently; 2 families with de novo DDEB out of 126 families with DEB in the Spanish cohort. The risk of recurrence in future siblings of the proband, this is not 50%, as is the case with inherited mutations, but estimated to be approximately 2% to 5%,52 given the possibility that one of the parents has a mosaic mutation in the germline. Of note is that the risk of recurrence in children of the proband is indeed 50%. In the case of patients with de novo dominant forms, the advice given by the expert clinical geneticist is essential.
We should also highlight that there is a tendency among physicians to make a genetic diagnosis of suspicion of de novo dominant transmission in patients with mild dystrophic forms and unaffected parents. Given the low frequency of appearance of de novo dominant mutations in the Col VII gene, this type of diagnosis should be avoided until the genetic result is obtained and the mode of transmission determined. This is because, in most cases, these patients actually have recessive mutations that result in a mild clinical presentation.
Autosomal Recessive Epidermolysis BullosaWhen a case of DEB appears for the first time in a family, we tend to think in terms of autosomal recessive transmission in which both gene copies (paternal and maternal) need to be mutated for the disease to become manifest. In these cases, each progenitor is a healthy carrier (heterozygotes) of one of the 2 mutations present in the DNA of their affected child. The family risk of new cases is 25% for successive siblings of the patient. In these cases, healthy siblings of affected individuals should undergo genetic study as well when they reach adulthood to determine whether they are carriers or not. It is important to point out that carriers can only have affected children if their partner is also a carrier. The probability that, by chance, the partner is a carrier is less than 1 in 50 (2%), unless there is a consanguineous relationship (or the partner is from a nearby region with a higher prevalence of an EB-associated mutation). Once the mutation has been identified in the carrier, it is possible to screen for this gene in the partner to rule out their status as a carrier with a reasonable degree of certainty.
This information, regarding the pattern of transmission, and its reproductive implications, including the options described below for prenatal and preimplantation diagnosis, among others, must be provided in a visit known as genetic counselling or advice. This type of visits are regulated and defined by the Spanish law of Biomedical Research ([LIB] 14/2007); and the services provided by the Spanish national health system are set in the Order SSI/1356/2015, dated July 2. Genetic counselling or advice is defined as the procedure intended to inform a person about the possible consequences (for themselves and their offspring) of the results of a genetic analysis or screening and the benefits and risks and, where appropriate, the advice given regarding possible alternatives derived from the analysis. Counselling may occur both before and after a genetic test or screening and even if such tests or screening did not take place. This advice should be provided in a nondirective fashion and complying with the principles of beneficence and nonmaleficence so that patients can make own decision.
The approach is common for any type of genetic or hereditary disease, and is applied in the same way in EB. Some recommendations on advising patients with EB and their families have been published,52,53 although the application of new techniques requires to review their applicability and scope.
Prenatal DiagnosisOnce a mutation has been detected in an individual affected by EB, prenatal diagnosis is technically feasible.54 In this case, the partner is attended in a visit, where information on the reproductive options available, as well as the obstetric techniques used to access the DNA of the fetus (from cells present in the amniotic fluid or chorionic villi), diagnostic possibilities, and risks or complications of these invasive techniques, is provided. Prenatal diagnosis can only be made if the gene responsible for EB and the mutation/s in that particular family are known.
In addition in some types of EB, other techniques can help support and reach prenatal diagnosis in specific cases. For example, detection of increased alfa-fetoprotein in amniotic fluid along with polyhydramnios during pregnancy are grounds for suspicion of a specific form of EB associated with pyloric atresia (EB-PA), which is detectable by ultrasound during the fetal period55. EB-PA, oftenly fatal, is caused by defects in α6β4 integrins or in plectin.22,40 On the other hand, in the first trimester, chorionic villi clearly express both proteins and their expression persists throughout pregnancy. Thus, chorionic villus analysis by immunofluorescence is a tool for prenatal diagnosis of EB-PA.56 Unfortunately, the fact that increased alfa-fetoprotein and polyhydramnios are nonspecific symptoms in pregnancy that may indicate other diseases together with the low casuistic of EB-PA, obstetricians may not associate these symptoms with a skin fragility disease in the absence of family history.
One of the novelties of the field is noninvasive prenatal diagnosis based on study of fetal DNA found in maternal plasma. The main advantage over conventional prenatal diagnosis is that there is no risk for the fetus or mother since a maternal blood sample is just required. Noninvasive prenatal diagnosis of monogenic diseases is only performed in research context and for some mutations. In the maternal plasma sample, fetal DNA coexists with much higher quantities of DNA of maternal origin. The coexistence of the 2 DNAs in the sample limits these studies to analysis of regions of the fetal DNA that are not present in the mother¿s genome that is, they have been inherited de novo or from the father. Despite this limitation, this noninvasive method has been shown to be effective for detecting mutations.57
Preimplantation DiagnosisCurrently, assisted reproduction techniques offer the possibility of diagnosis of embryos and their subsequent implantation in the maternal uterus if they are free of the disease-causing mutations. This is known as preimplantation genetic diagnosis (PGD). It is recommended that couples turn to an Assisted Reproduction Service experienced in the management of this type of diagnosis, work in coordination with a Genetics Service and have optimized in vitro fertilization protocols, generally extensive, in order to achieve an adequate number of embryos to analyze. PGD is available in Spain after its approval of Law 14/2006, dated May 26, on techniques of assisted human reproduction, offers an alternative reproductive option for families in the case of severe hereditary diseases with early onset for which no curative treatment is currently available, such as EB. It is recommended that couples attend to an assisted reproduction service with expertice in handling this type of diagnosis, in coordination with a genetic department. Such a service should have optimized in vitro fertilization protocols (usually extensive) in order to obtain sufficient embryos for analysis.
The first premise for performing PGD is the availability of a precise genetic diagnosis for the couple, that is, that the specific mutations carried by both partners have been identified. An informative genetic study of the couple is also required to select the most appropriate genetic markers. That is, study of genetic markers that accompany the mutation should allow differentiation of the haplotype that is associated with the disease. These markers are analyzed in this particular couple to estimate the possible combinations in future embryos. In this way, it can be determined a priori whether the combination of markers of the 2 progenitors will enable to distinguish beyond any doubt which are affected embryos and which are healthy ones. If the study of genetic markers is noninformative, that is, that the combinations of markers make it impossible to distinguish between affected and healthy embryos, PGD might not be technically feasible, depending on the subsequent diagnostic strategy.
In the event that the couple is suitable for the study, a protocol similar to that of a conventional in vitro fertilization with intracytoplasmic sperm injection must be followed, although in the case of PGD, a higher number of ovules are required. For this, the future mother should follow a pharmacological treatment of controlled ovarian stimulation, consisting of injection of stimulating hormones, with the aim of ensuring that several ovules mature in the same cycle. After extraction of the ovules (mature follicles), in vitro fertilization is carried out. Three days after fertilization, when the embryos are formed of 6 to 8 cells (onset of the morula stage), a cell (blastomere) is biopsied from each embryo and sent to the genetics laboratory for diagnosis to determine which embryos carry the disease and which do not (healthy embryos). This study should take no longer than 24hours.
Of the embryos considered healthy for the mutation and, therefore, transferable, it is necessary to select once again which of these have the best morphological characteristics for transfer to the maternal uterus (never more than 3 at a time) and which thus have the greatest chance of success. Given that more than one embryo is transferred, more than 1 may become implanted, leading to a multiple pregnancy. If the process should fail for whatever reason, the woman can start the process once again with a new cycle of ovarian stimulation.
During the informed consent process, the couple should be informed of their expected particular results and of the success and failure rates. They should then be kept up to date with the results obtained during the whole procedure. For example, they should know that the pregnancy rate with these techniques is low (less than 20% in each attempted cycle). PGD has a reliability between 88% and 96%,58 and so conventional prenatal diagnosis is recommended once pregnancy is established to confirm the results.
In Spain, prenatal genetic diagnosis and preimplantation diagnosis are included in the services offered by the national health service when the established criteria for genetic risk, characteristics of offspring, and fertility study and age of the couple are met. Therefore, the procedure is free through the public health service for couples who are carriers of EB when they meet these criteria. Each autonomous community has a reference center available for preimplantation genetic study. Couples should consult the corresponding official body in their respective autonomous community. This process is included in the range of services of the national health service, applicable throughout Spain. It is regulated by the Human Assisted Reproduction law and by the Commission for Human Assisted Reproduction for situations that are not contemplated by the law.
FundingDr. del Río¿s group and, in particular, the molecular diagnostics, was financed by specific funds from CIEMAT and IIS-FJD, the following granted projects CELLCAM, GENEGRAFT HEALTH-F2-2011-261392, SAF2013-43475-R, ICI14/00363, CIBERER INTRAMURAL/08/714.1, the Fundación Ramón Areces (CIVP-16A1864) and the Patient Associations, Berritxuak and DEBRA-España. The group of Dr. Ayuso thanks the resources obtained from PIE13/00051, CIBERER CB06/07/0036, and Biobanco PT13/0010/0012.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We would like to thank individuals with EB and their families for their constant trust, respect, and willingness to cooperate with our group.
We also thank the Asociación Piel de Mariposa (Butterfly Skin Association) DEBRA-España.
Likewise we express our gratitude to the health care professionals such as nurses, geneticists, dermatologists, pediatricians, surgeons, and other specialists who, in some way, have participated in molecular diagnosis; without their collaboration and dedication this article would not have been possible. In particular, we also thank our most regular collaborators for their time to facilitate contact with patients, sampling and clinical information (including some of the clinical photos that illustrate this article). We also acknowledge the groups of Dr. Meneguzzi (INSERM-Francia) and Dr. Zambruno (Istituto Dermopatico dell’Immacolata, currently, Ospedale Pediatrico Bambino Gesù-Italia), world leaders in the field of EB for their support, especially at the beginning.
Finally, we thank the EB diagnostic team, Nuria Illera, Almudena Holguín, Dr. Adela García, Dr. Marta García, Dr. Álvaro Meana, Dr. Sara Llames, Eva García, and Dr. Ángeles Mencía for their excellent work and dedication.
The collaboration between the groups of Dr. Carmen Ayuso and Dr. Marcela del Río has been essential for transmitting the molecular diagnosis to the patient through genetic counselling and prenatal diagnosis.
Cátedra Fundación Jiménez Díaz de Medicina Regenerativa y Bioingeniería Tisular: http://www.fjd.es/iis_fjd/es/areas-grupos-investigacion/tecnologia-innovacion-sanitaria/grupos-asociados/medicina-regenerativa-ingenieria-tejidos.
Spanish Epidermolysis Bullosa association. www.pieldemariposa.es/
Health care professionals: Dr. Raúl de Lucas, Dr. Rocío Maseda, and Dr. Juan Carlos López (Hospital La Paz, Madrid); Dr. Ángela Hernández and Dr. Antonio Torrelo (Hospital Niño Jesús, Madrid); Dr. José Lasso (Hospital Universitario Gregorio Marañón, Madrid); Dr. Asunción Vicente and Dr. Loreto Martorell (Hospital St Joan de Deu, Barcelona); Dr. Pilar Iranzo, Dr. Susana Puig, and Dr. José Manuel Mascaró (Hospital Clínic, Barcelona); Dr. Raquel Rodríguez and Dr. Isabel Febrer (Consorcio Hospital General, Valencia); Dr. Agustí Toll and Dr. Ramón Pujol (Hospital del Mar, Barcelona); Dr. Yolanda Gilaberte (Hospital San Jorge, Huesca); Dr. Juan Luis Santiago (Hospital Ciudad Real); Dr. José Bernabeu (Hospital Virgen del Rocío, Seville); Dr. Ángel Vera (Hospital Carlos Haya, Malaga); Dr. Eulalia Baselga (Hospital Santa Creu i Sant Pau, Barcelona); Dr. Sebastián Mir-Mir and Dr. Roger Costa (Hospital Plató de Barcelona); Dr. Raquel Sáez (Hospital Universitario de Donostia, San Sebastián); Dr. Elena Arana (Hospital Vall d’Hebron, Barcelona); and Dr. José Carlos Moreno (Hospital Reina Sofía, Cordoba).
Please cite this article as: Sánchez-Jimeno C, Escámez MJ, Ayuso C, Trujillo-Tiebas MJ, del Río M, en representación de la Cátedra de la Fundación Jiménez Díaz de Medicina Regenerativa y Bioingeniería Tisular, et al. Diagnóstico genético de la epidermólisis bullosa: recomendaciones de un grupo español de expertos. Actas Dermosifiliogr. 2018;109:104–122.
The names of all the health professionals are listed in Appendix A. C. Sánchez-Jimeno and M.J. Escámez are both first authors: they have contributed equally to this paper.
articles



www.publicationethics.org.
