

Nodular mucinosis is a chronic primary idiopathic mucinosis that, according to the 2001 classification proposed by Rongioletti et al.,1 belongs to the group of the localized forms of lichen myxedematosus, which includes acral persistent papular mucinosis, a mild form of papular mucinosis, self-healing papular mucinosis, papular mucinosis of infancy, and the nodular form. Localized mucinoses are characterized by the appearance of small numbers of waxy papules (or plaques or nodules due to confluence), usually on the lower limbs or trunk. These localized forms must be differentiated from the diffuse form or scleromyxedema, which is characterized by a rash of groups of small, firm waxy papules of around 2 to 3mm in diameter, arising predominantly on the upper part of the trunk and on the neck, face, forearms, and hands.2 The papules show a linear distribution and the perilesional skin is shiny. Muscle, joint, nervous system, gastrointestinal, pulmonary, or otohinolaryngologic manifestations may be present, and paraproteinemia is an almost constant feature. Atypical and intermediate forms also exist; these include patients with scleromyxedema but with no systemic signs or paraproteinemia, patients with localized forms associated with paraproteinemia, and combined or not otherwise specified cases.1
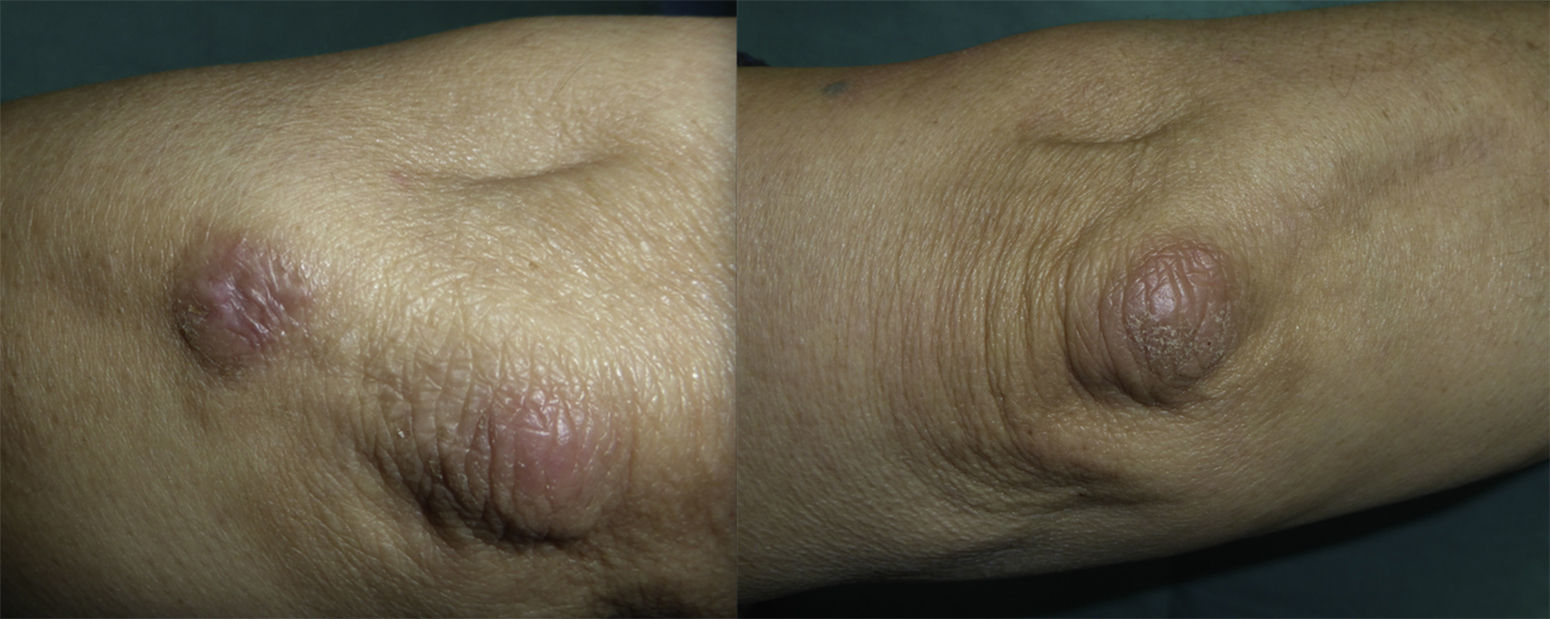
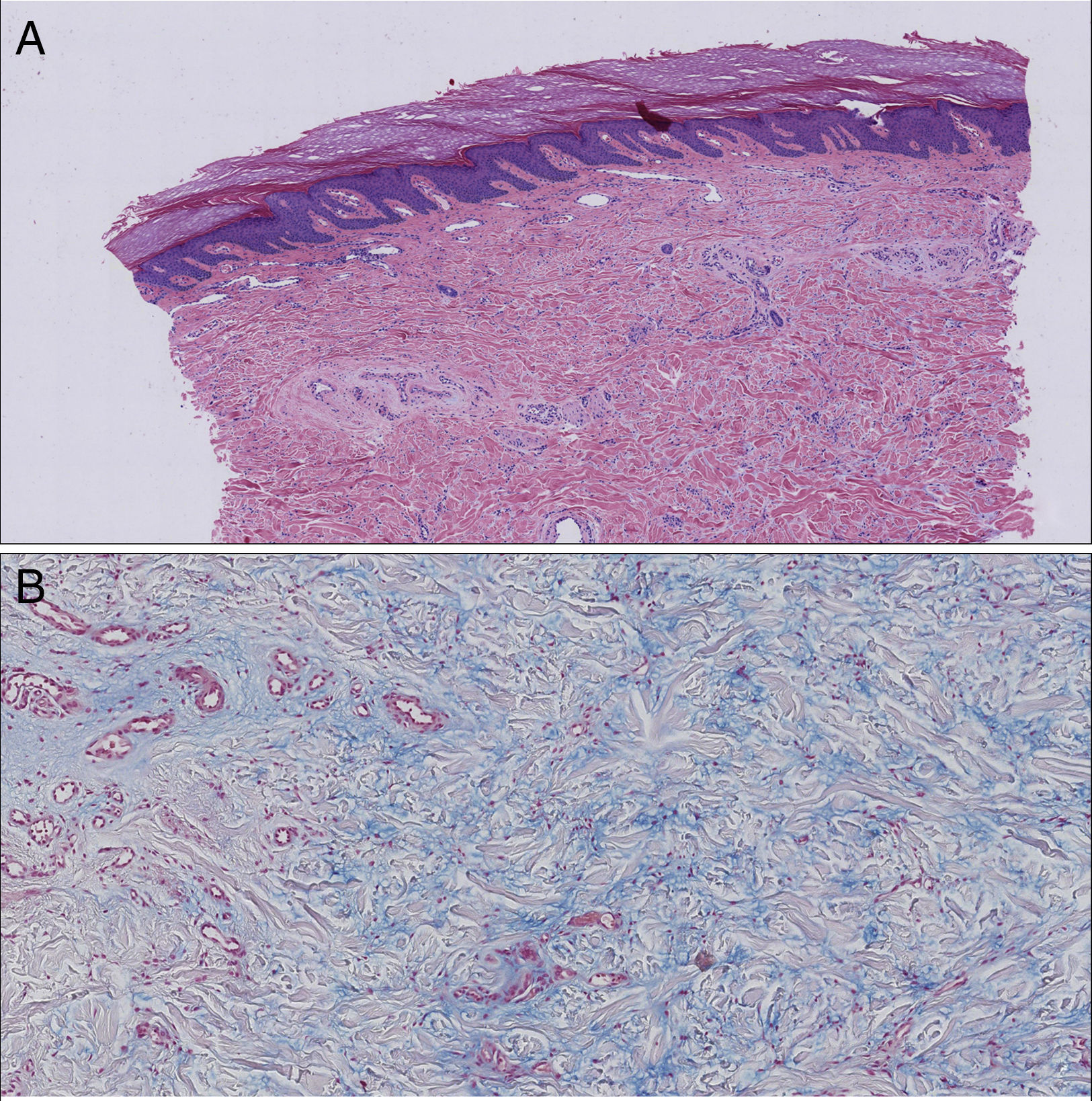
We present the case of a 72-year-old woman with no past history of interest. She was seen in our department for the appearance 3 months earlier of 2 brownish erythematous plaques with a slightly scaly surface on both elbows (Fig. 1). The lesions had grown both radially and in thickness and were slightly tender. Biopsy revealed mild orthokeratotic hyperkeratosis with papillomatosis and, in the papillary dermis, abundant interstitial mucin deposits that stained positive with alcian blue. The diagnosis was localized mucinosis (Fig. 2).


Laboratory tests including complete blood count, biochemistry, and 24-hour urine analysis were normal. Plasma protein electrophoresis showed a peak of 890mg/dL in the gamma region that was shown by immunofixation to be an oligoclonal band in a polyclonal background. Serum free light chains were measured and a kappa chain of 30mg/L was detected, with a kappa-to-lambda ratio of 1.88. The hematology department did not consider it necessary to perform bone marrow study. With these results, we made a diagnosis of light chain monoclonal gammopathy of uncertain significance based on the Myeloma Working Group 2014 criteria.3 Treatment was started with topical clobetasol propionate under occlusive dressings for 1 month, with almost complete resolution of the lesions. At the time of writing, the patient continues on follow-up in the hematology department.
This was therefore an atypical form of lichen myxedematosus; only 4 such cases have been published to date.4–7 The pathogenesis of the association is unknown. In 1 of the cases, the skin lesions disappeared 8 years after their onset and the paraproteinemia some years later, with no systemic clinical repercussions.4 Another case occurred in a 60-year-old may with skin lesions clinically and histologically compatible with papular mucinosis. After 3 months of treatment with oral corticosteroids with no response, the patient presented disorientation and a deterioration in his general state that led to a diagnosis of immunoglobulin M type multiple myeloma. He then received standard therapy with bortezomib and dexamethasone and the skin lesions disappeared after 4 treatment cycles.6 Finally, the interesting case of a 38-year-old woman with lesions of papular mucinosis that presented a Köebner phenomenon—a feature not previously described in the literature—and a monoclonal gammopathy of uncertain significance.7
It should be noted that, as previously mentioned by other authors,1 the terms lichen myxedematosus, scleromyxedema, and papular mucinosis have tended to be used interchangeably in the literature and in daily clinical practice and, despite the 2001 reclassification,1 confusion between the terms persists, particularly regarding the atypical forms such as ours.
In our patient, thanks to the cutaneous manifestations, we will continue to monitor her closely and, if there is progression to light chain multiple myeloma (estimated incidence of 0.3% per year3) or to primary amyloidosis, it should be possible to make an early diagnosis and initiate treatment, which is fundamental to the prognosis of this disease.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Rodríguez-Jiménez P, Chicharro P, Ascensión A, de Argila D, Daudén E. Mucinosis nodular asociada a gammapatía monoclonal de cadenas ligeras de significado incierto. Actas Dermosifiliogr. 2017;108:272–273.



