


Neurofibromatosis type 2 is an autosomal dominant hereditary disease with complete penetrance. It gives rise to multiple central and peripheral nervous system tumors, ocular alterations, and various types of skin lesion. In general, neither dermatologists nor other specialists have in-depth knowledge of the clinical manifestations of neurofibromatosis type 2. In some cases, this can lead to delayed diagnosis, which can increase morbidity and mortality. We describe the less well known clinical manifestations of NF2, focusing particularly on skin lesions specific to this disease. Identification of these lesions, when present, can facilitate diagnosis.
La neurofibromatosis tipo 2 es una enfermedad hereditaria, autosómica dominante, con penetrancia completa, que ocasiona la aparición de múltiples tumores en el sistema nervioso central y periférico, afectación ocular y lesiones cutáneas de distinta índole. La clínica de la neurofibromatosis tipo 2 es, en general, poco conocida, tanto por los dermatólogos como por el resto de los especialistas, lo que deriva, en algunos casos, en un retraso en el diagnóstico que favorece un aumento de la morbilidad y la mortalidad. En este artículo se expondrán las manifestaciones clínicas menos conocidas, haciendo especial hincapié en las lesiones dermatológicas propias de la enfermedad, las cuales en caso de presentarse y ser identificadas, pueden facilitar el diagnóstico de la misma.
Neurofibromatosis type 2 (NF2) is an autosomal dominant hereditary disease with complete penetrance. It gives rise to multiple central and peripheral nervous system tumors, ocular disorders, and various types of skin lesions. There is no predilection for sex or race and, although initially considered a very uncommon disease, recent studies suggest that it has an incidence close to 1/25 000 patient-years and a prevalence of 1/60 000 patients.1–3
Unlike neurofibromatosis type 1, which is caused by a mutation in the neurofibromin gene (located on chromosome 17q11.2), NF2 is the result of mutations in the NF2 gene located on the long arm of chromosome 22 (22q12.2). This gene encodes a protein known as schwannomin or merlin, which comprises 595 amino acids. The molecule is so named because of its resemblance to the ezrin, radixin, and moesin proteins (merlin is in fact an acronym for moesin-ezrin-radizin-like protein).2,4 These proteins are present in vertebrates, and their primary function is to bind cytoskeletal proteins (such as actin) with membrane glycoproteins.2,4 Merlin is also promotes such binding and, in addition, is able to regulate cell proliferation through this same binding (probably due to contact-mediated growth inhibition phenomena).2 It is strongly expressed in nervous tissue and, for this reason, most tumors occurring in NF2 are derived from neural cells.2 When merlin has mutated, inhibition of cell division does not occur effectively, thus favoring the appearance of tumors of neural origin and the clinical manifestations characteristic of NF2. The condition is not widely known, either among dermatologists or other specialists, and so diagnosis may be delayed in some cases, with the corresponding increase in morbidity and mortality of the disease. In addition, half of the cases of NF2 arise due to spontaneous mutations in patients without a family history, further hindering the diagnostic process.1,5
In this article, the clinical manifestations of the disease are presented, with particular emphasis on skin lesions. If present and properly identified, these lesions can facilitate early diagnosis of the disease and determine the prognosis of the patient.
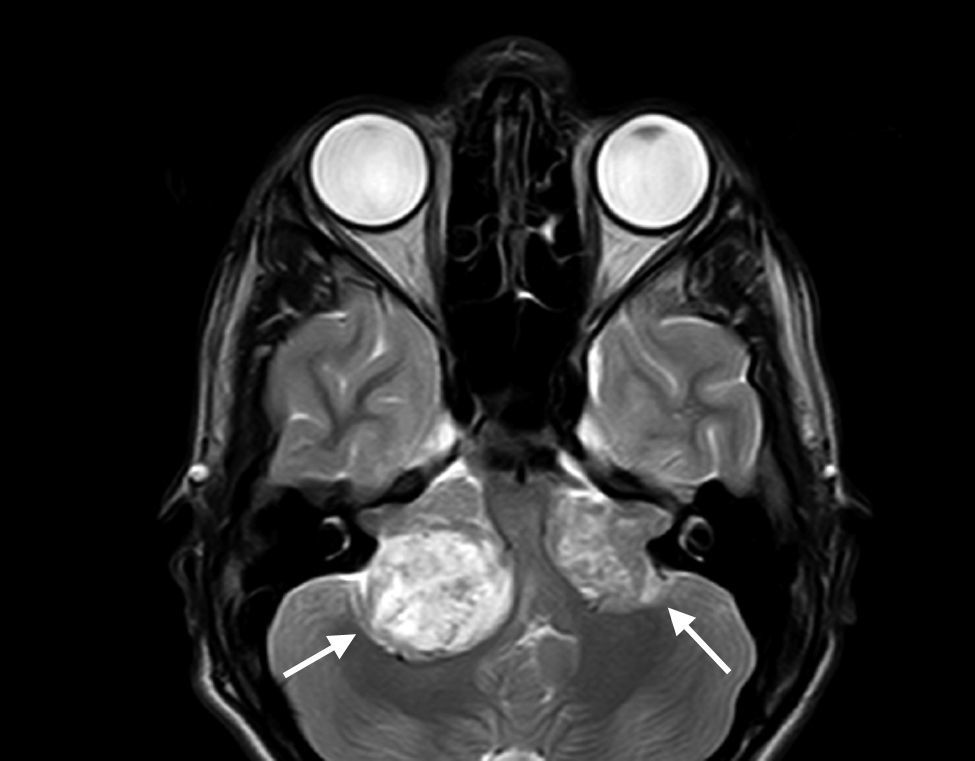
Neurological ManifestationsThe most widely-known characteristic of NF2 is the development of bilateral vestibular schwannomas (Figure 1).3 The presence of such lesions leads to diagnosis of the disease in the majority of cases and is considered a strong enough criterion for diagnosis (Table 1). These tumors appear in 95% of the patients and cause neurosensory deafness that presents at 27 years of age on average.2 Hearing loss is present in 95% of patients (although only two-thirds are aware of this) and does not depend on tumor diameter but rather the degree of nerve compression. In most patients with NF2, this is the first sign of the process.2 In the most serious cases, however, NF2symptoms present at a younger age and usually with less recognizable clinical manifestations, such as the onset of tumors at other sites, onset of neurological symptoms, and presence of skin or ocular lesions.1,4 One example of this is the appearance of schwannomas in other nerves of the body. Approximately 60% of those affected by NF2 have schwannomas in nerves other than cranial nerve 8,6 with the trigeminal nerve the second most frequently involved cranial nerve (29% of patients).2,5 These tumors can also be formed in other anatomical areas such as the spine. This occurs in 90% of patients with the disease, although only 30% of them have symptoms.5,6
 in the magnetic resonance imaging of a patient with neurofibromatosis type 2.")
Diagnostic Criteria for Neurofibromatosis Type 2 (NF2).
| Diagnostic criteria for NF2 |
|---|
| Bilateral vestibular schwannomas |
| First-degree family member with NF2+ |
| Unilateral vestibular schwannoma or |
| Two of the following: meningioma, schwannoma, glioma, neurofibroma, or cataracts (posterior subcapsular) |
| Unilateral schwannoma+ |
| Two of the following: meningioma, schwannoma, glioma, neurofibroma, or cataracts (posterior subcapsular) |
| Multiple meningiomas+ |
| Unilateral schwannoma or |
| Two of the following: schwannoma, glioma, neurofibroma, or cataracts (posterior subcapsular) |
In addition to schwannomas, NF2 can produce tumors derived from other neural cells such as meningiomas, ependymomas, and astrocytomas.1–4 Of all these, meningiomas are the most relevant. These are diagnosed in half the patients with NF2 and publications that assessed mortality associated with the disease concluded that these lesions are one of the main prognostic markers.7 They grow faster than schwannomas, and clinically, they are manifest as headaches or seizures (if they form within the cranium) or muscle weakness, pain, and paresthesia (if they form in the spine).8
Ependymomas, for their part, appear in the spine, are usually low grade and grow slowly.1–4 Astrocytomas, in contrast, usually form in the brainstem.1–4
Unlike in neurofibromatosis type 1, the risk of malignant conversion of tumors is low in NF2. The probability that a central nervous system tumor undergoes malignant transformation is approximately 0.5%.8
NF2 can also be expressed in the form of mononeuropathy that leads to progressive loss of function of the affected nerves, leading in turn to muscle deficit in the region that they innervate. This form of involvement is seen in 10% to 15% of cases, with the facial nerve the one most often affected.2 In children, this is a frequent manifestation, although the relationship with the disease is often not recognized at the time of presentation unless there is a family history of NF2. In this case, detailed examination of the skin can be key to suspect diagnosis because, as discussed later (see Skin Manifestations section), in this context, skin lesions specific to the disease are often observed. In exceptional cases, serious polyneuropathy, the origin of which is not entirely clear, has even been reported. The phenomenon is, however, uncommon as it is only observed in 3% to 5% of cases.1,9,10
Skin ManifestationsSkin lesions are not included in the criteria for diagnosis of the disease because it is generally thought that, unlike in neurofibromatosis type 1, skin manifestations in NF2 are uncommon and not very specific. However, nothing could be further from the truth; skin lesions in NF2, although not as frequent as in neurofibromatosis type 1, can be sufficiently characteristic to cause suspicion of the disease and even confirm diagnosis.
Essentially, 2 types of skin lesions can be identified in this context: café-au-lait spots and cutaneous schwannomas, with the latter lesions being the most characteristic.1,4
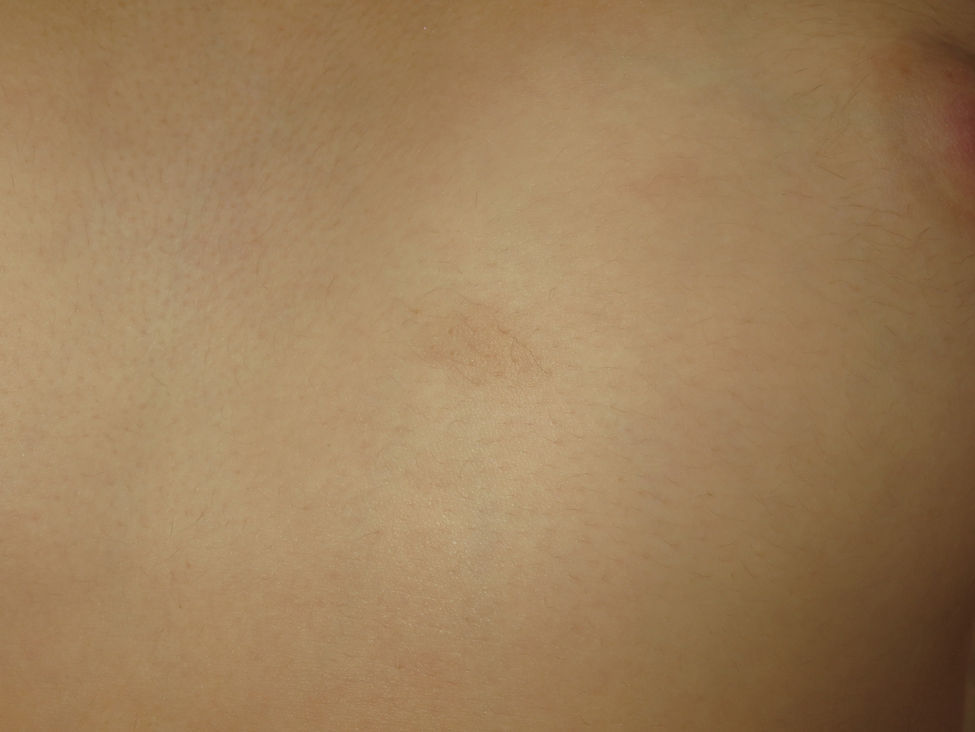
Café-au-lait spots are observed in 40% of patients1,2 and they have a similar but not identical morphology to those of neurofibromatosis type 1. In NF2, these lesions are less frequent (fewer than 1% of patients with NF2 have 6 or more café-au-lait spots), have a lighter color, and have more irregular borders (Figure 2).4 Their histology is very similar to those of the typical lesions of neurofibromatosis type 1, but with fewer melanosomes.4,5 In patients with manifestations in the first years of life, the presence of these lesions is more frequent and can be detected in almost all patients in some pediatric series.4

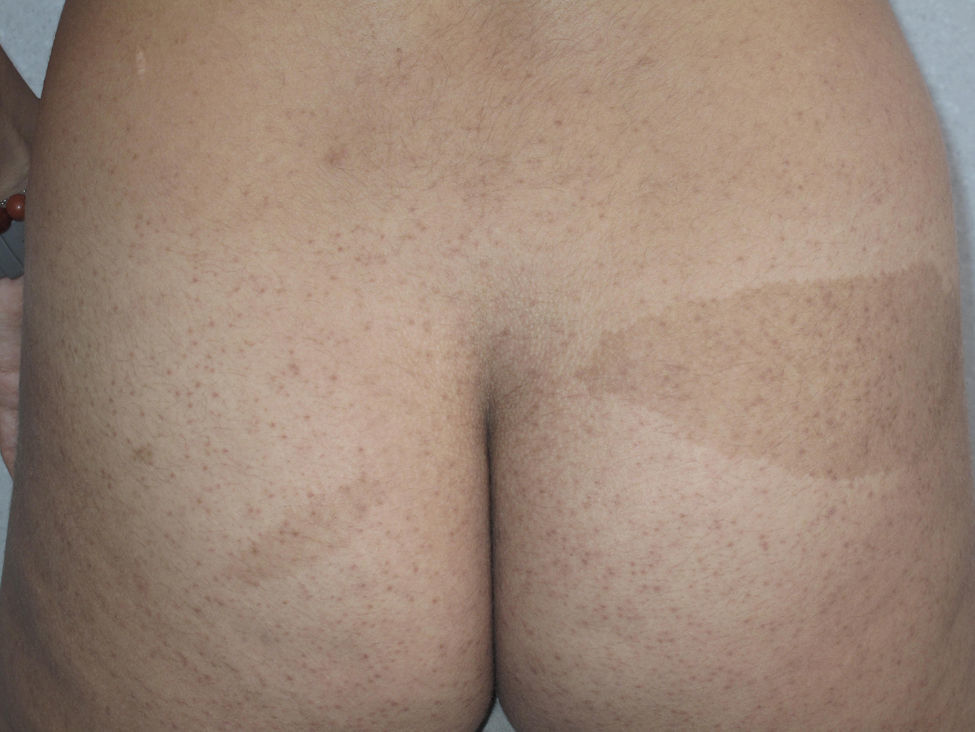
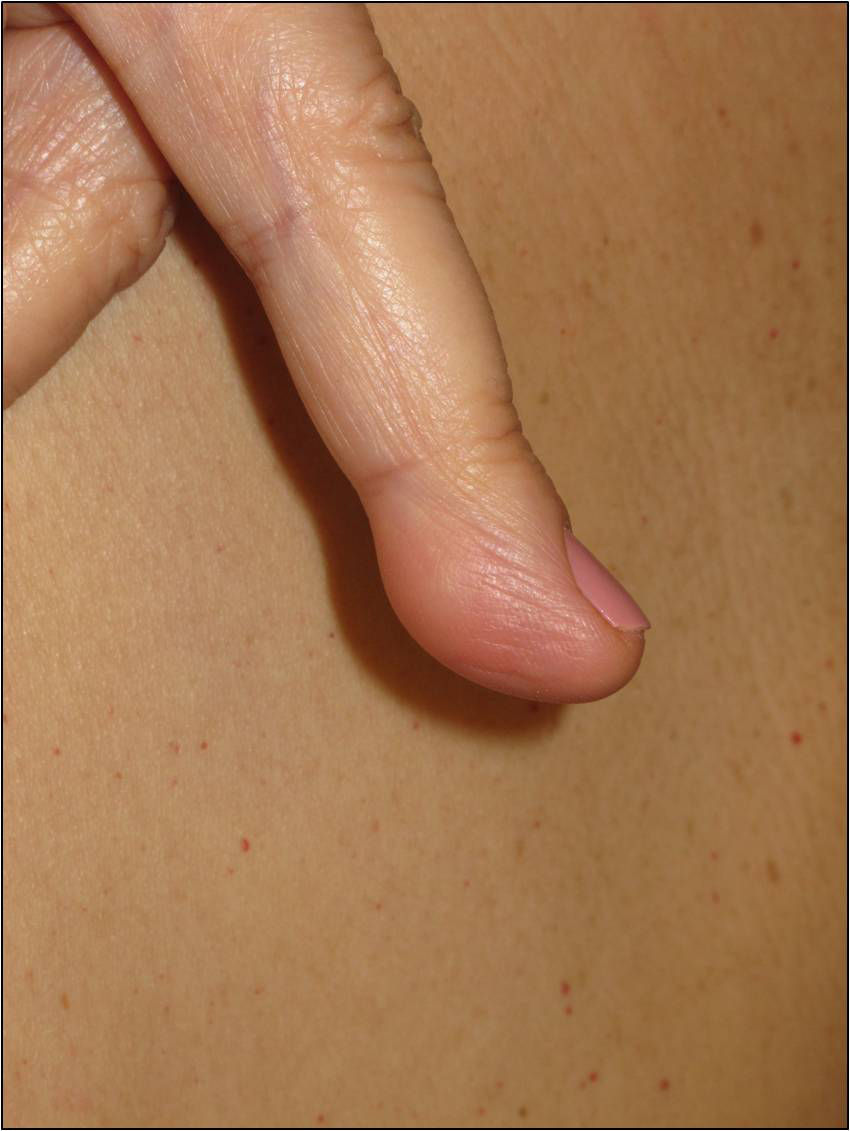
Cutaneous schwannomas can take on different clinical forms depending on the depth at which they develop: if they form in the dermis they give rise to superficial lesions identified in the literature as plaques11 but if they develop at a deeper site their clinical presentation is more that of a nodule or tumor.11 It is safe to say that that the most extensively characterized lesions are plaques, which correspond clinically to areas of mildly hyperpigmented skin that is soft to touch with occasional hypertrichosis. They are preferentially located on the chest or abdomen (Figures 3 and 4).1 These lesions are observed in 41% to 48% of patients and are the ones with the earliest onset.4,5,11 In the most severe forms of the disease, as occurs with café-au-lait spots, the presence of these lesions is more common.4,5


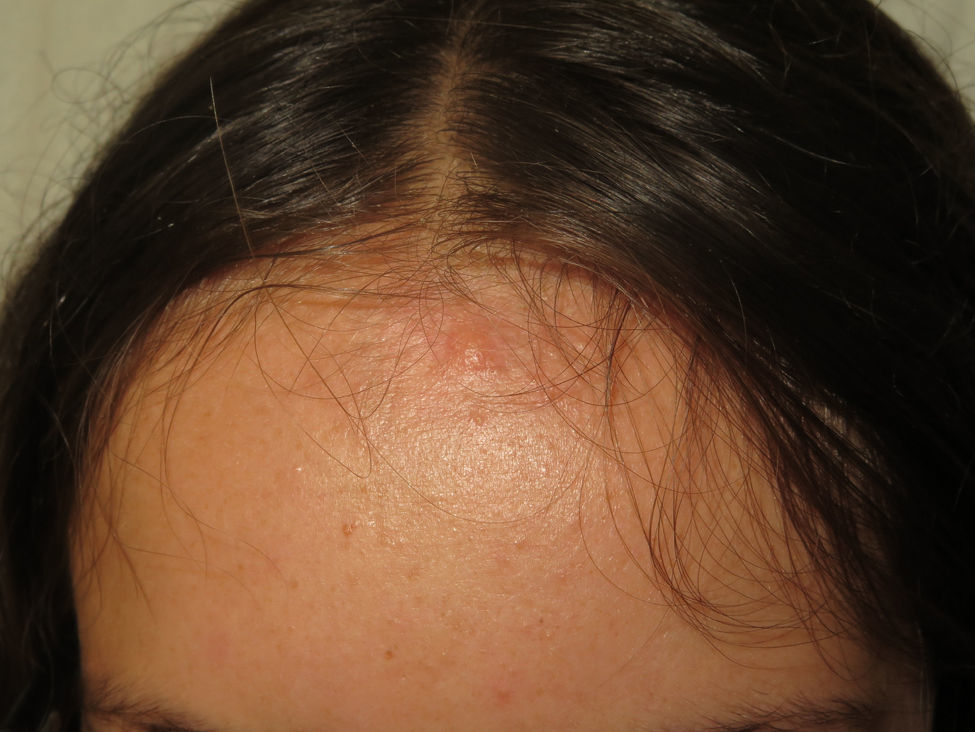
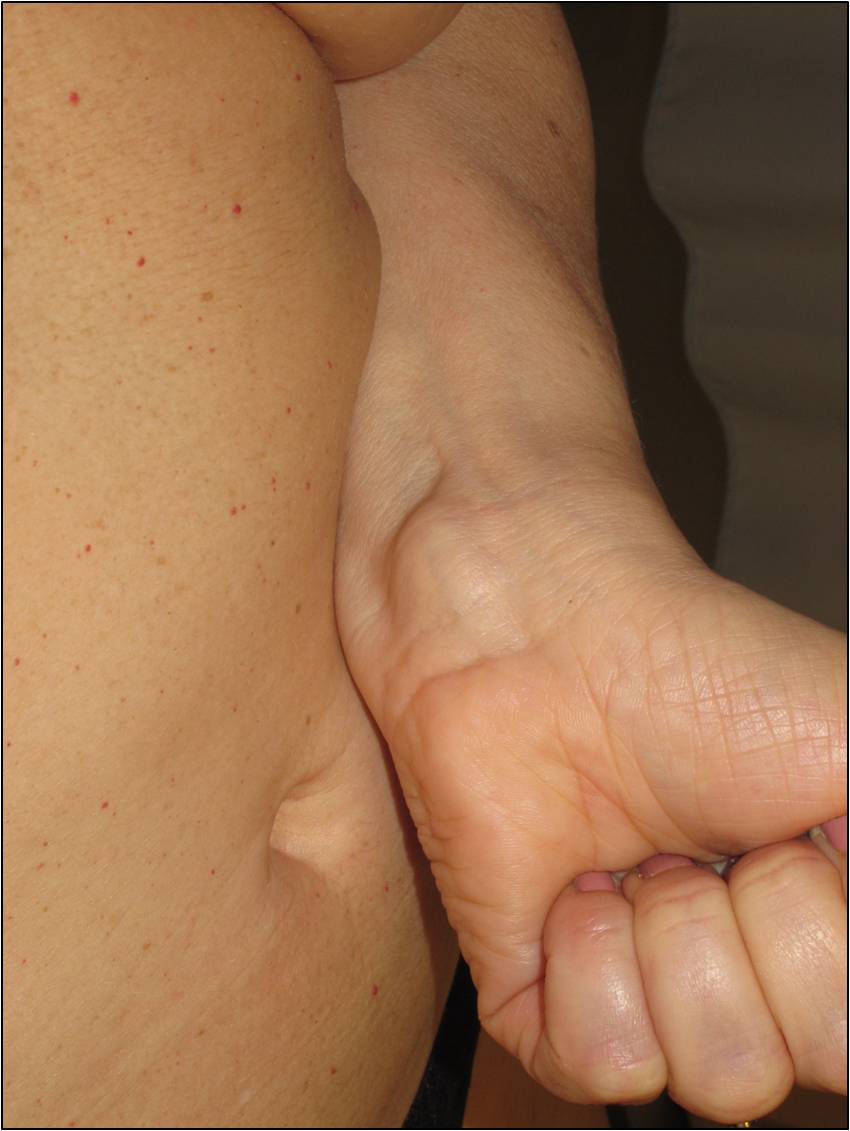
The nodules or tumors are covered with normal skin and are more palpable than visible (Figures 5 and 6). The most profound lesions have an elastic consistency and can be painful to manipulation and follow paths of innervation. When these lesions are more superficial, in contrast, they feel softer.1,2 In some series, the presence of cutaneous schwannomas in children with NF2 is seen in up to 90% of patients,4 although the number of lesions per patient might not be very high (only 10% of patients have more than 10 tumors).1


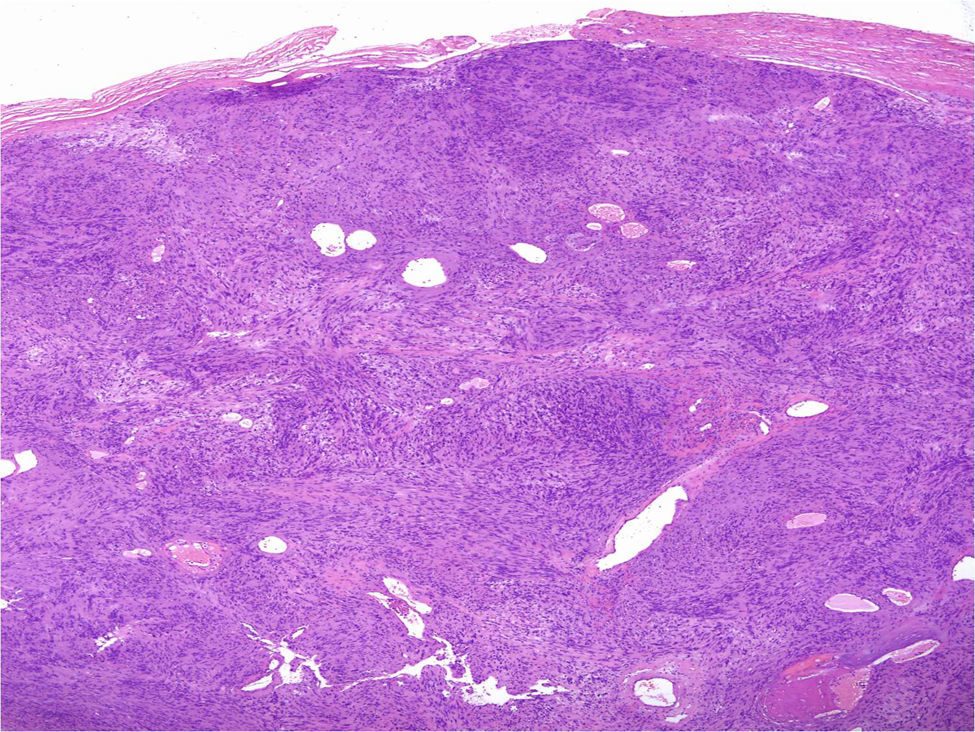
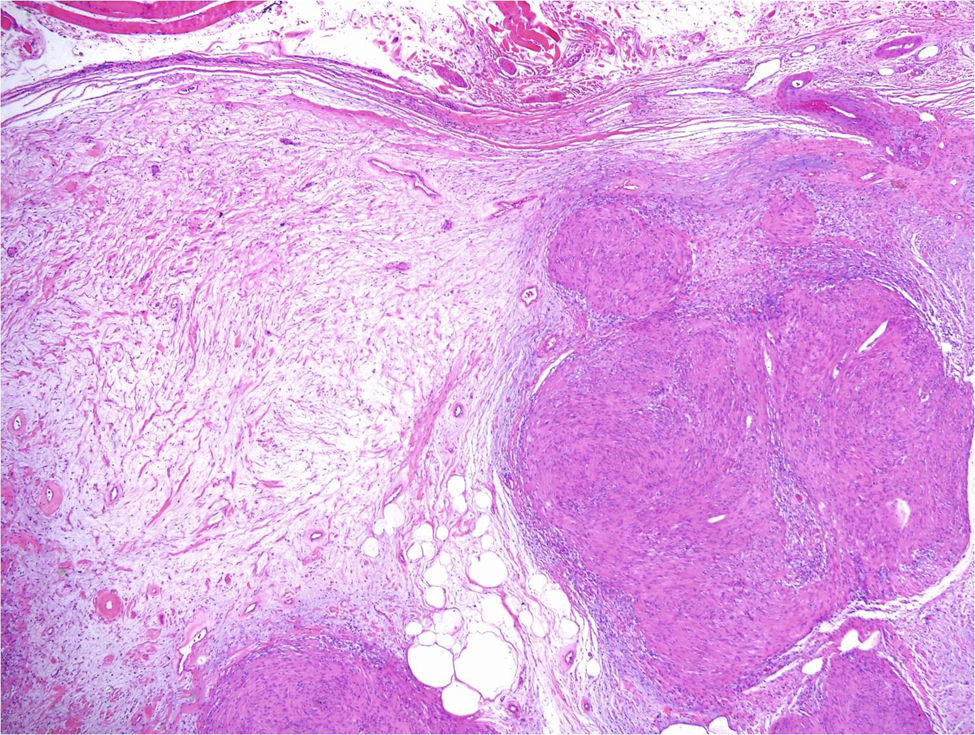
Under the microscope, different patterns can be observed for cutaneous schwannomas. The most widely known, according to the literature, is the classic schwannoma.8,12 This type is made up of a spindle-cell proliferation, located in the dermis. The lesion is compact and encapsulated, with typical Antoni A areas rich in cells (in which Verocay bodies can occasionally be seen) and Antoni B areas with lower cellularity. The cells of these proliferations are stained with S100 and vimentin (Figure 7).8,12

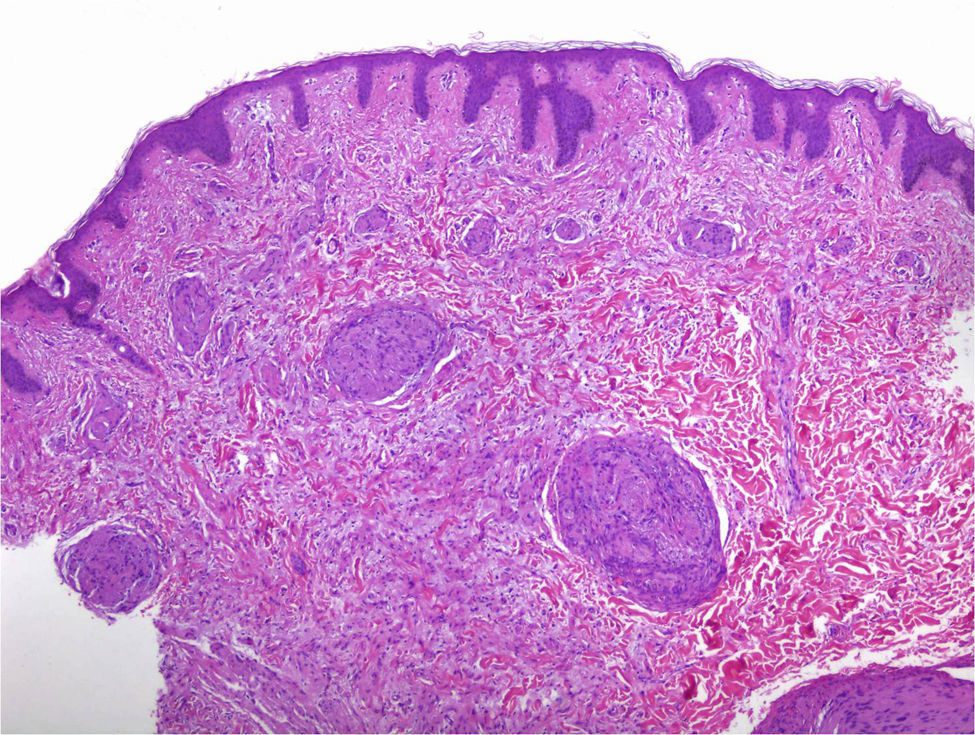
In addition to the classic pattern, schwannomas with a plexiform pattern13 and hybrid tumors12 have also been identified. In schwannomas with a plexiform pattern, bundles of very compact spindle-cells have been observed (Antoni type A), which spread along the dermis forming bundles of nervous tissue to give the plexiform appearance (Figure 8).13 In hybrid tumors, diffuse areas of neurofibroma are observed within which classic schwannoma nodules are located (Figure 9).12 Hybrid tumors are uncommon and have been reported both in the context of neurofibromatosis type 1 and NF2. Their significance is not well known. Immunohistochemistry of hybrid tumors is the same as for the 2 types of constituent tumor. That is, all schwannoma cells are stained with S100 whereas only 40% of those in areas of neurofibroma are stained. In addition, other typical findings are apparent in areas of neurofibroma, such as for example axons stained for neurofilaments or fibroblasts positive for factor XIIIa.12


There are no studies that report in sufficient detail the clinical correlation of each of the microscopic patterns, but what is certain is that the plaques and most superficial tumors translate, from a microscopic point of view, into a schwannoma with plexiform pattern or a hybrid tumor, whereas the deeper tumors that follow the path of the nerve probably have the nodular microscopic pattern of classic schwannoma.
The formation of cutaneous schwannomas is very characteristic of NF2 but not exclusive. The literature has reported cases of patients with solitary lesions with the classic schwannoma pattern, plexiform schwannoma,14 and even a hybrid tumor15 under the microscope. In addition, these lesions can also be seen in schwannomatosis, and less frequently, in some cases of neurofibromatosis type 1.14,15 Schwannomatosis is an entity caused by mutations in the IN11/SMARCB1 gene. This gene is located on chromosome 22 in close proximity to the NF2 gene. Mutation of IN11/SMARCB1 leads to the formation of multiple schwannomas in different areas of the body (intracranial, spinal, or peripheral nerves) as well as in the skin and soft tissues, but without the typical vestibular schwannomas characteristic of NF2.14
In addition to café-au-lait spots and schwannomas, in an isolated case published by Spanish authors, the presence of hypopigmented lesions has also been reported.16
Ophthalmological ManifestationsAmong the most frequent ophthalmological manifestations are cataracts, which are seen in between 60% and 80% of cases if a rigorous ophthalmological examination is undertaken.1 These lesions are almost always located in a posterior subcapsular site. The development of hamartomas in the retina or meningiomas in the optic nerve has also been reported.1
Characteristics of Neurofibromatosis Type 2 in Pediatric PatientsAs mentioned above, clinical expression of NF2 in children differs in many aspects from expression in adults. In children, there have been reports of a high proportion of schwannomas in nerves other than cranial nerve 8 (in fact, only 15% to 30% of children present with vestibular schwannomas), a higher proportion of meningiomas located in the spine, a higher frequency of mononeuropathy, and in some patients even intracranial calcifications similar to those seen in some cases of tuberous sclerosis.4,5 Moreover, as mentioned above, skin manifestations in this group of the population are particularly predominant. Bearing in mind that the most superficial schwannomas, which manifest as small plaques, are the earliest lesions to present, these are also the lesions seen most frequently. Thus, detailed skin examination may be worthwhile in a child with facial paralysis to detect skin lesions suggestive of schwannoma. If such lesions are present, the disease may be diagnosed.
It is also important to point out that cataracts and retinal hamartomas in children may be a very frequent finding (although there are large variations between series).4 As these alterations are often asymptomatic (only 20% of patients with ocular lesions present with decreased visual acuity),4,5 ophthalmologic assessment should be requested in all children in whom this process is suspected. These patients may also have strabismus if the oculomotor nerves are affected by tumors in these structures.4
Segmentary FormNF2 can also present in segmentary form when mosaicism occurs. It is estimated that among all patients with spontaneous appearance of NF2 (50% of all patients), 25% to 35% present with a mosaic form.17 In these cases, the clinical manifestations are mild and schwannomas and skin lesions can be limited to specific anatomical parts of the body (for example, to one half of the body) and not generalized. In these patients, differential diagnosis with schwannomatosis is complicated because vestibular schwannomas do not usually present and molecular diagnostic techniques are normally required.18
Early DiagnosisAll the above manifestations contribute to a high morbidity and mortality associated with NF2. Some series report mean survival after diagnosis of 15 years.5 Early diagnosis is essential to improve survival. This is useful for several reasons. First, it facilitates the detection of vestibular tumors while they are still small, thus enabling planning of the different therapeutic options available5 and it also gives the patients the chance to learn sign language while they are still able to hear (thus facilitating adaptation to the disease).5 Finally, it means that patients can be referred quickly to specialist reference centers. This last point is one of the most important. In fact, studies that have attempted to identify prognostic factors have demonstrated that the fact that attendance of a reference center led to statistically significant improvement in survival.19
These centers are able to offer a multidisciplinary approach and surgeons more experienced in these types of tumors. This is also of utmost importance given these are uncommon tumors, and the experience of the surgeon is vital to preserve the facial nerve and hearing, as well as to reduce postoperative complications.8 This all contributes to an improved quality of life for patients with NF2, and even leads to decreased mortality. In Spain, the 2 reference centers available are Hospital Sant Joan de Déu for pediatric patients and the Hospital Germans Trias i Pujo for adult patients.
Early diagnosis is even more important when NF2 presents in the early years of life. It has been shown that when the first symptoms of NF2 appear during childhood, the disease has a worse prognosis and mortality is higher.4 It is calculated that 1 in 5 patients with NF2 will present with the first signs of the disease before the age of 10 years.4 In series that include pediatric patients, it has been shown that the first manifestations are not usually vestibular schwannomas as is the case in adults but rather neurological or skin disorders.4,5 Dermatologists therefore have a crucial role for suspecting and confirming diagnosis of the disease, even though other tumors in the central or peripheral nervous systems are not present.
One of the challenging clinical aspects of NF2, both for patients and clinicians, is phenotypic variability, that is, the diversity of clinical manifestations, as well as the different ages of onset of symptoms. On the one hand, there is marked variation within families. Traditionally, 2 forms of NF2 are distinguished, a severe form (Wishart) characterized by a greater number of tumors, lower age of onset of NF2 symptoms, and higher prevalence of cataracts; and a milder form (Gardner) characterized by fewer tumors, older age of onset of symptoms, and lower prevalence of cataracts. Currently, a possible genotype-phenotype correlation has been reported whereby patients with mutations resulting in a truncated protein (nonsense or frameshift mutations) have more severe disease than patients with missense mutations, somatic mosaicism, or mutations that result in complete loss of protein product (large deletions).20
The presence of skin lesions in the early phases of the more serious forms of the disease, in particular the most superficial schwannomas that are manifest in form of small plaques, which as we have seen are very characteristic and accessible for study, mean that identification of these lesions has a high diagnostic yield that will benefit the patient.
Molecular DiagnosisWhen the disease is suspected at early ages, patients do not usually meet all of the necessary clinical criteria for diagnosis. In such cases, molecular diagnostic techniques are used. Some patients may also request genetic counselling.
The NF2 gene consists of 17 exons. For molecular diagnosis, it is necessary to rule out the presence of isolated pathogenic point mutations or deletions and partial or total duplications of the gene. To achieve this, a study is performed that begins with taking a blood sample from the patient for short-term lymphocyte culture to obtain DNA and RNA. Subsequently, the region encoding the NF2 gene is amplified by polymerase chain reaction of RNA (cDNA) and all amplification products are analyzed by direct sequencing in search of point mutations. If a mutation is detected, this is confirmed by PCR and sequencing of the exon involved. If a point mutation is not observed, the presence of deletions or partial or total duplications of the NF2 gene should be ruled out by multiplex ligation-dependent probe amplification. The sensitivity of these techniques is 91% to 95%8,11 if the mutation is present in the majority of cells.
In cases of mosaicism, this process is more complex because the mutation is not usually present in the patient's blood. In such cases, the genetic mutations should be sought in at least 2 tumors.17
ConclusionsIn summary, NF2 is an uncommon disease that is associated with a high morbidity and mortality, above all when onset occurs during the first years of life. Early diagnosis of the process is a priority, as this improves survival and quality of life of the individuals affected. NF2 with onset in the early years of life can be diagnosed from skin, ocular, and neural lesions, but not so much from the manifestations arising from vestibular schwannomas. For this reason, greater clinical and microscopic understanding of the skin lesions of NF2 is key for improving the chance of early diagnosis, with a corresponding improved prognosis.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Plana-Pla A, Bielsa-Marsol I, Carrato-Moñino C, en representación del grupo de Centros, Servicios y Unidades de Referencia (CSUR) en Factomatosis. Manifestaciones cutáneas de la neurofibromatosis tipo 2: interés diagnóstico y pronóstico. Actas Dermosifiliogr. 2017;108:630–636.



