

Multiple cerebral cavernous malformations (MCCMs) is characterized by the presence of multiple vascular malformations in the central nervous system. It can also affect the retina and the skin. The disease usually presents sporadically; of the patients that present the familial form, the majority show autosomal dominant inheritance.1 We describe 2 families with familial MCCMs diagnosed because of skin lesions detected in 2 children of 12 and 13 years of age.
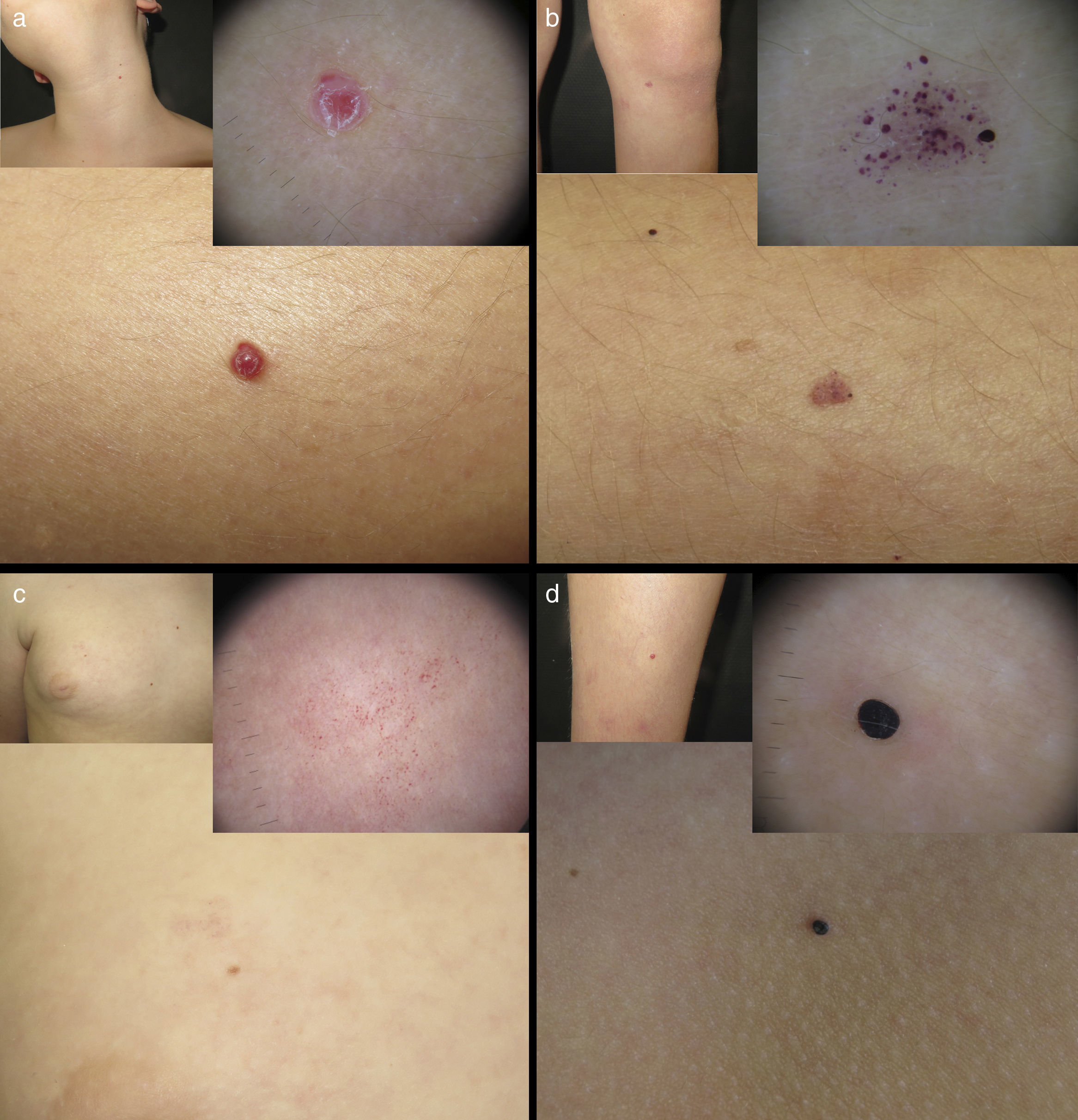
Our first patient was a 12-year-old boy with no past history of interest. He was seen for asymptomatic skin lesions of vascular appearance that had been present for a year. Physical examination revealed 2 reddish papules similar to cherry angiomas on the neck and right leg (Fig. 1A), 3 reddish-pink macules with a dotted dermoscopic pattern on the trunk and left leg (Fig. 1B), an erythematous macule on the trunk, with a reticulated vascular dermoscopic pattern (Fig. 1C), and 2 small, violaceous angiokeratoma-like papules on the trunk and left leg (Fig. 1D). The initial family history was negative, but a targeted medical history revealed the presence of cerebrovascular lesions in the maternal grandmother, diagnosed on magnetic resonance imaging (MRI) performed 5 years earlier for sudden onset hearing loss. Skin examination of the grandmother revealed multiple reddish, cherry angioma-type papules on the trunk. The medical history and physical examination of the other family members only detected 2 bluish nodular lesions suggestive of venous malformations on the right arm of the patient's mother. Given the family history, MRIs of the patient and his mother were requested, observing the presence of cerebral cavernous malformations in both of them. Genetic analysis was performed on the 3 affected family members, revealing the c.268C>T mutation, a substitution of arginine that gives rise to a stop codon at position 90 of exon 6 of gene KRIT1 (CCM1) (Molecular Genetics Laboratory, Hôpital Lariboisière, Paris, France), which confirmed the diagnosis of MCCMs.

Distinct vascular lesions in our first patient. A, Cherry angioma on the right leg, with vascular lacunae and desquamating collarettes in the dermoscopic image. B, Capillary malformation on the left leg with a dotted vascular dermoscopic pattern. C, Capillary malformation with a reticulated vascular dermoscopic pattern. D, Angiokeratoma-like lesion with a dark lacuna in the dermoscopic image.
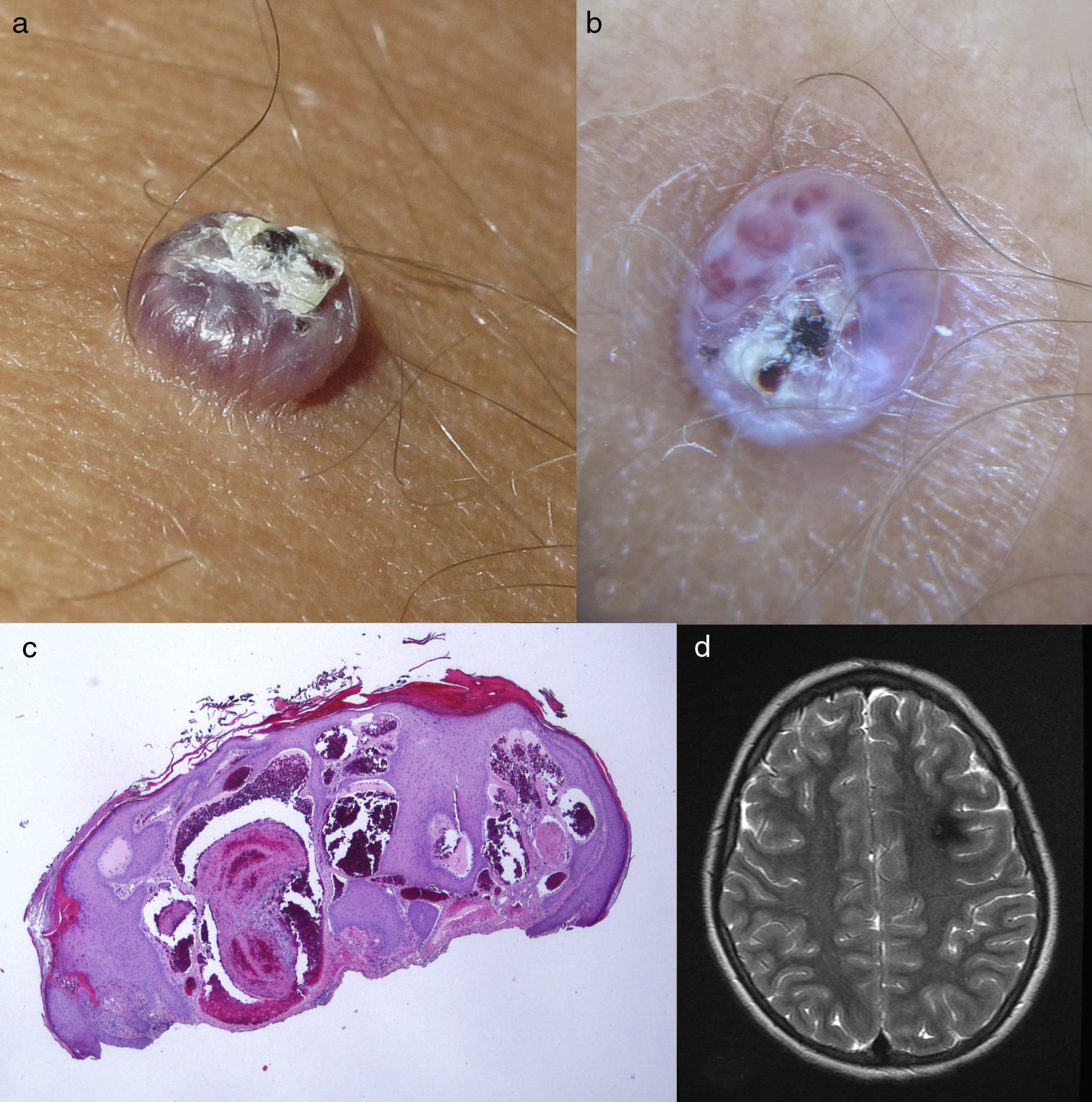
The second patient was a 13 year-old boy with no past history of interest. He presented a lesion that had arisen on his knee some months earlier. The clinical manifestations, dermoscopy, and histology were suggestive of angiokeratoma (Fig. 2, A-C). The medical history revealed that the father had been diagnosed with MCCMs. The patient's MRI revealed MCCMs (Fig. 2D). The genetic analysis showed a pattern of deletion of exons 1, 2, and 3 of gene KRIT1 (CCM1) (Genetics Department, Fundación Jiménez Díaz, Madrid, Spain).

A, Angiokeratoma on the knee. B, Dermoscopic image with vascular lacunae and a superficial scab. C, Histology showing vessels with fibrin thrombi in the dermis and epidermal hyperplasia. Hematoxylin and eosin, original magnification×4. D, Axial T2-weighted cerebral magnetic resonance image: cerebral cavernous malformation.
MCCMs can arise sporadically (80%) or be familial (20%).2 The familial form has an autosomal dominant pattern of inheritance with variable clinical penetrance. To date, 3 responsible genes have been identified—CCM1 (KRIT1), CCM2 (MGC4607), and CCM3 (PDCD10)—with more than 100 different mutations.3 The KRIT1 gene, detected in both our families, is the most common mutation in patients with skin lesions.4,5 The etiology and pathogenic mechanism are unknown, although it has been observed that the 3 proteins coded by the responsible genes (KRIT1, MGC4607, and PDCD10) are implicated in angiogenesis and in vascular remodeling.6
In MCCMs, the cerebral lesions remain asymptomatic in up to 40% of patients.7 Clinical manifestations include epileptic crises, headaches, and focal neurological deficits, due either to hemorrhage or to the compression of adjacent structures. The diagnosis is based on imaging studies, preferentially MRI.7 No action protocols or guidelines have been drawn up to specify the monitoring and follow-up of these patients.
In 2009, Sirvente et al.4 presented the longest series of patients with MCCMs, with 417 cases, and reported skin involvement in 9%. Those authors described 3 types of vascular skin involvement: hyperkeratotic capillary-venous malformation, which is the most common (39%) and specific; capillary malformation (34%); and venous malformation (21%). Furthermore, they described 2 types of capillary malformations: punctate capillary malformation (most common) and port wine stain capillary malformation. The majority of patients in that series presented solitary vascular skin lesions, which can make the diagnosis difficult to suspect clinically if there is no family history of the disease.
In conclusion, we have presented 2 families with MCCMs and skin involvement. Our first patient had multiple and varied vascular lesions, whereas the second had a single lesion, an angiokeratoma.
When these cutaneous vascular lesions are detected, especially if they are multiple or in pediatric patients, it is important to take a detailed targeted medical history to detect any history of skin or cerebral lesions in relatives and to consider to performing cerebral MRI or molecular studies. Additional studies allow central nervous system lesions to be detected early to be able to plan the therapeutic strategy and rapid intervention in patients who develop neurological symptoms.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Escudero-Góngora MM, Bauzá A, Giacaman A, Martín-Santiago A. Cavernomatosis cerebral múltiple: cuando la clave del diagnóstico está en la piel. Actas Dermosifiliogr. 2017;108:680–683.



