

In recent decades, an association has been reported between epidermolysis bullosa (EB) and dilated cardiomyopathy (DC). DC is typically in an advanced phase when detected, leading to a poorer prognosis. Our objective was to determine the prevalence of DC in patients with EB seen in Hospital San Joan de Déu in Barcelona, Spain, between May 1986 and April 2015.
MethodsThis was a descriptive, cross-sectional chart-review study in which we recorded the type and main subtypes of EB and the presence or absence of DC.
ResultsFifty-seven patients with EB were found, 19 with EB simplex, 10 with junctional EB, 27 with dystrophic EB (14 dominant dystrophic and 13 recessive dystrophic), and just 1 with Kindler syndrome. DC was detected in only 2 patients with recessive dystrophic EB. Twenty-three patients had presented factors that could have had a causal relationship with the potential onset of DC.
ConclusionsDC is a possible complication of EB, particularly in recessive dystrophic EB. Periodic follow-up should be performed to make an early diagnosis and start treatment.
En las últimas décadas se ha descrito la asociación entre epidermólisis ampollosa (EA) y miocardiopatía dilatada (MD). Generalmente esta última enfermedad se detecta en fases avanzadas, implicando un peor pronóstico.
Nuestro objetivo consistió en determinar la prevalencia de MD en los pacientes con EA vistos en el Hospital San Joan de Déu (Barcelona) desde mayo de 1986 a abril de 2015.
MétodosEstudio descriptivo transversal mediante revisión de las historias clínicas con atención al tipo y subtipos mayores de EA y la existencia o no de MD.
ResultadosSe recogieron 57 pacientes con diagnóstico de EA. De ellos 19 presentaban EA simple, 10 EA juntural, 27 EA distrófica (14 EA distrófica dominante y 13 EA distrófica recesiva) y existió un caso de síndrome de Kindler. Solo 2 de los pacientes con EA distrófica recesiva presentaron MD. En 23 de los pacientes con EA existieron factores que podrían tener una relación causal con el potencial desarrollo de MD.
ConclusiónLa MD puede ser una complicación en los pacientes con EA, mayoritariamente del subtipo de EA distrófica recesiva, por lo que deben hacerse controles periódicos para su temprano diagnóstico y tratamiento.
There are 3 main subtypes of cardiomyopathy: dilated, hypertrophic, and restrictive. Dilated cardiomyopathy (DCM) is defined as progressive dilation of the heart with contractile dysfunction of the left or the left and right ventricles.1 One characteristic of DCM is congestive heart failure.2 DCM is uncommon in the pediatric population, with an incidence of 0.6 to 1.2 cases per 10 000 children.1,2 Diverse factors have been implicated in its etiology, such as idiopathic, familial or genetic, viral, and autoimmune factors, micronutrient deficiency, iron overload, chronic anemia, and toxic or pharmacological agents.1,2
Inherited epidermolysis bullosa, which we will refer to throughout this article as epidermolysis bullosa (EB), comprises a group of genetic disorders characterized by skin fragility and blistering. These disorders as classified as EB simplex, junctional EB (Herlitz and non-Herlitz), dystrophic EB (dominant or recessive), and Kindler syndrome. The classification of EB according to the 2008 consensus report is shown in Table 1.3,4
Current Classification of Inherited Epidermolysis Bullosa (EB).
| EB simplex (EBS) | Suprabasal | Basal |
| Lethal acantholytic EB Plakophilin syndrome EBS superficialis | EBS, localized EBS, Dowling-Meara or herpetiform EBS, generalized (non-Dowling-Meara) EBS with mottled pigmentation EBS with pyloric atresia EBS, Ogna EBS, migratory circinate | |
| Junctional EB (JEB) | Herlitz type | Non-Herlitz type |
| JEB, non-Herlitz, generalized JEB with pyloric atresia JEB, inversa JEB, late-onset Laryngo-onycho-cutaneous syndrome | ||
| Dystrophic EB | Dominant dystrophic EB (DDEB) | Recessive dystrophic EB (RDEB) |
| DDEB DDEB, acral DDEB, pretibial DDEB, pruriginosa DDEB, nails only DDEB, bullous dermolysis of the newborn | RDEB, severe generalized RDEB, generalized other RDEB, inversa RDEB, pretibial RDEB, pruriginosa RDEB, centripetalis RDEB, bullous dermolysis of the newborn | |
| Mixed EB | Kindler Syndrome |
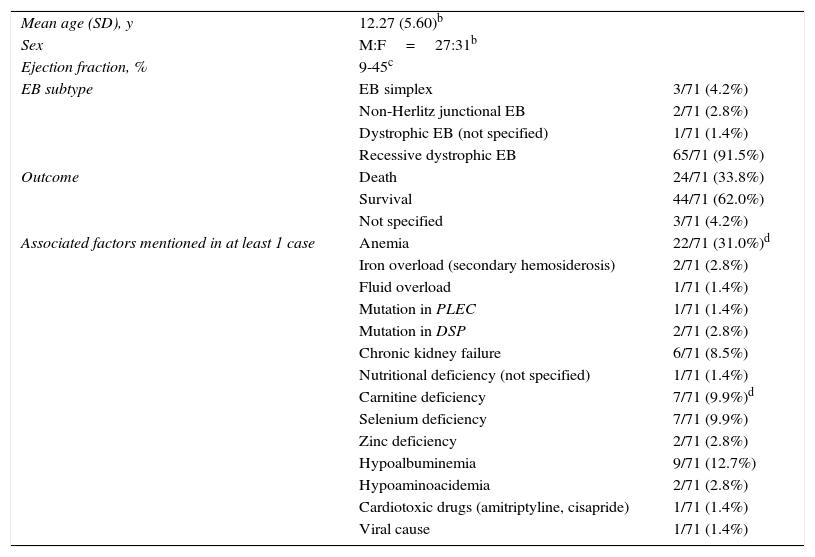
An association between EB and DCM has been observed in recent decades, particularly in patients with recessive dystrophic EB. The cases published to date are summarized in Table 22,5–14 and additional details are provided in Supplementary Table 1.
Summary of Epidermolysis Bullosa (EB) Cases Associated With Dilated Cardiomyopathy in the Literaturea
| Mean age (SD), y | 12.27 (5.60)b | |
| Sex | M:F=27:31b | |
| Ejection fraction, % | 9-45c | |
| EB subtype | EB simplex | 3/71 (4.2%) |
| Non-Herlitz junctional EB | 2/71 (2.8%) | |
| Dystrophic EB (not specified) | 1/71 (1.4%) | |
| Recessive dystrophic EB | 65/71 (91.5%) | |
| Outcome | Death | 24/71 (33.8%) |
| Survival | 44/71 (62.0%) | |
| Not specified | 3/71 (4.2%) | |
| Associated factors mentioned in at least 1 case | Anemia | 22/71 (31.0%)d |
| Iron overload (secondary hemosiderosis) | 2/71 (2.8%) | |
| Fluid overload | 1/71 (1.4%) | |
| Mutation in PLEC | 1/71 (1.4%) | |
| Mutation in DSP | 2/71 (2.8%) | |
| Chronic kidney failure | 6/71 (8.5%) | |
| Nutritional deficiency (not specified) | 1/71 (1.4%) | |
| Carnitine deficiency | 7/71 (9.9%)d | |
| Selenium deficiency | 7/71 (9.9%) | |
| Zinc deficiency | 2/71 (2.8%) | |
| Hypoalbuminemia | 9/71 (12.7%) | |
| Hypoaminoacidemia | 2/71 (2.8%) | |
| Cardiotoxic drugs (amitriptyline, cisapride) | 1/71 (1.4%) | |
| Viral cause | 1/71 (1.4%) | |
Abbreviations: F, female; M, male.
The aim of this study was to determine the prevalence of DCM in pediatric patients with EB.
MethodsWe performed a descriptive cross-sectional study of all cases of EB seen at Hospital de San Joan de Déu, a pediatric referral hospital in Barcelona, Spain, between May 1986 and April 2015. Patients with a clinical diagnosis of EB were identified through a database providing access to medical records and photographs. Based on the consensus recommendations for the classification of EB,3 patients were divided into the different EB subtypes based on clinical findings and test results (standard histology, antigen mapping, electron microscopy, genetic studies). Information was also noted on reasons for referral to cardiology, presence or absence of echocardiographic alterations, presence or absence of DCM (diagnosed by a pediatric cardiologist and defined as ventricular dilation and reduction in systolic function), and laboratory findings possibly associated with DCM. All the data were recorded retrospectively and based only on information contained in the patients’ charts (history, physical examination, and other tests).
ResultsSeventy-two patients with a diagnosis of EB were identified through the database. After exclusion of those with a doubtful diagnosis or lacking the test results required for correct classification, 57 patients were included. Nineteen had EB simplex, 10 had junctional EB (4 Herlitz and 6 non-Herlitz), 27 had dystrophic EB (14 dominant and 13 recessive), and 1 had Kindler syndrome.
Cardiological and echocardiographic assessment had been performed in 19 patients (5 with EB simplex, 1 with Herlitz junctional EB, 4 with non-Herlitz junctional EB, 2 with dominant dystrophic EB, and 7 with recessive dystrophic EB). Among them, 17 patients showed no signs or symptoms of cardiac involvement and had been referred to the cardiologist to investigate early DCM. Two patients had been referred with a clinical suspicion of cardiomyopathy (confirmed in 1 case).
DCM was diagnosed in just 2 patients, both with recessive dystrophic EB. This corresponds to a prevalence of 3.5% (2 of the 57 children analyzed). The prevalence of DCM by EB subtype was 0% for EB simplex, junctional EB, and Kindler syndrome (0/19, 0/10, and 0/1, respectively) and 7.4% (2/27) for dystrophic EB. Considering only the patients with recessive dystrophic EB, the prevalence of DCM was 15.4% (2/13).
In the first of the 2 cases in which a diagnosis of DCM was confirmed, the condition was detected when the patient was 13 years old due to signs and symptoms of heart failure and compatible echocardiographic findings (ventricular dilation and ejection fraction [EF] <50%). The patient was treated with carvedilol at doses adjusted to symptoms and EF. Initially, the EF remained at around 50%, but it decreased significantly (to 20%) during flares. Four years after diagnosis of DCM, the patient showed progressive worsening and developed kidney failure due to systemic AA amyloidosis. He died at the age of 21 years, with severely deteriorated general health and multiple organ failure. Factors that may have contributed to the development of DCM are chronic anemia, multiple regular blood transfusions, hypoalbuminemia, hypoaminoacidemia, and zinc and selenium deficiency.
The second patient was diagnosed with DCM during a routine echocardiographic examination when she was 3 years of age. She has since been receiving treatment with carvedilol (2mg/12h). The EF has remained at around 45% and there have been no signs of heart failure. Factors that could have contributed to the development of DCM in this patient include anemia, regular blood transfusions, hypoalbuminemia, hypoaminoacidemia, and carnitine deficiency.
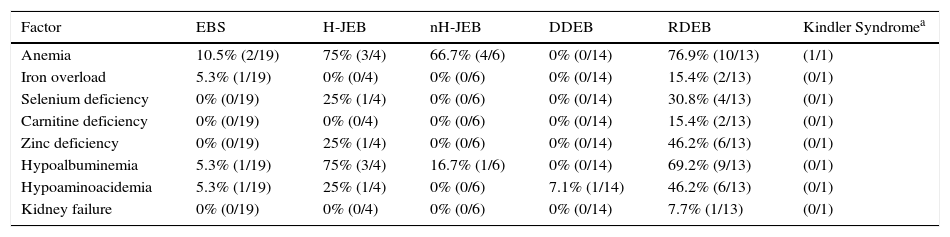
Table 3 shows the presence of factors potentially associated with the development of DCM in the patients in our series grouped by EB subtype. Twenty-three patients had at least 1 alteration that may have an etiological role in DCM. Eleven of these patients (48%) had recessive dystrophic EB.
Factors That Could Play a Role in the Development of Dilated Cardiomyopathy in the Patients in the Present Series.
| Factor | EBS | H-JEB | nH-JEB | DDEB | RDEB | Kindler Syndromea |
|---|---|---|---|---|---|---|
| Anemia | 10.5% (2/19) | 75% (3/4) | 66.7% (4/6) | 0% (0/14) | 76.9% (10/13) | (1/1) |
| Iron overload | 5.3% (1/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 15.4% (2/13) | (0/1) |
| Selenium deficiency | 0% (0/19) | 25% (1/4) | 0% (0/6) | 0% (0/14) | 30.8% (4/13) | (0/1) |
| Carnitine deficiency | 0% (0/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 15.4% (2/13) | (0/1) |
| Zinc deficiency | 0% (0/19) | 25% (1/4) | 0% (0/6) | 0% (0/14) | 46.2% (6/13) | (0/1) |
| Hypoalbuminemia | 5.3% (1/19) | 75% (3/4) | 16.7% (1/6) | 0% (0/14) | 69.2% (9/13) | (0/1) |
| Hypoaminoacidemia | 5.3% (1/19) | 25% (1/4) | 0% (0/6) | 7.1% (1/14) | 46.2% (6/13) | (0/1) |
| Kidney failure | 0% (0/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 7.7% (1/13) | (0/1) |
Abbreviations: DDEB, dominant dystrophic epidermolysis bullosa; EBS, epidermolysis bullosa simplex; H-JEB, Herlitz junctional epidermolysis bullosa; nH-JEB, non-Herlitz junctional epidermolysis bullosa; RDEB, recessive dystrophic epidermolysis bullosa.
An increased risk of heart disease has been observed in patients with EB. Fine et al.2 published a report on 15 patients with EB and congestive heart failure, and considered that most of the patients also had DCM. The subtypes associated with heart failure were recessive dystrophic EB and non-Herlitz junctional EB. The condition was most common in patients with severe generalized dystrophic recessive EB. The authors found the risk of congestive heart failure to be significantly greater in these patients than in the general population and even greater still in patients with concomitant chronic kidney failure. More recently, Ryan et al.14 investigated the presence of subclinical heart damage and other vascular disorders in patients with recessive dystrophic EB. They found that 40 of the 45 patients analyzed had echocardiographic abnormalities and that 18% had a dilated aortic arch.
The factors that have been most often linked to the development of cardiopathy in patients with EB are chronic anemia, iron overload, low carnitine and selenium levels, drugs, and concomitant viral infection.1
Chronic anemia generally has multiple causes. It responds poorly to dietary supplementation and requires regular blood transfusions. Both chronic anemia and iron overload secondary to repeated transfusions may have a role in the onset of DCM.1
In relation to nutritional deficiencies, EB patients with DCM have been found to have significantly lower levels of carnitine than those without.1 Selenium supplementation in cases of deficiency has been found to reduce morbidity and mortality in DCM. Nevertheless, no significant differences have been observed between selenium levels in patients with EB and DCM and patients with EB without DCM.8 Thiamine deficiency is another documented cause of DCM.8 Although there have been reports of DCM being reversed with restoration of carnitine levels8 and of improved symptoms with selenium or thiamine supplementation, micronutrient deficiency alone does not appear to be a causal factor in DCM, but it could be a contributory factor.1,13
Viral infections can also cause DCM.1 The heart damage in such cases is generally due to a postinfectious autoimmune process.8 Prodromal viral infection frequently goes unnoticed, as it can precede the onset of DCM by several months. Diagnosis is even more complicated in patients with aggressive forms of EB, who, for varying reasons, may exhibit symptoms such as fever or dyspnea of noncardiac origin.1
Cardiotoxic drugs can also exacerbate DCM in patients with EB and should therefore be withdrawn on detection of symptoms suggestive of cardiopathy.15
Apart from mutations in the PLEC1 gene12,16,17 (coding for plectin) in EB simplex with muscular dystrophy associated with DCM12,16,17 and mutations in the DSP gene (coding for desmoplakin) in lethal acantholytic EB,18,19 no other genetic alterations have been linked to the development of cardiomyopathy in EB. There is an interesting case of a patient with recessive dystrophic EB who had left ventricular noncompaction cardiomyopathy (LVNC) without DCM.20 The authors indicated that LVNC can progress to DCM in certain cases, and suggested that it might precede DCM in recessive dystrophic EB.
Patients with EB have an increased risk of DCM, and those who develop the disease have a mortality of between 30% and 60%. In patients with recessive dystrophic EB, DCM tends to be detected at around 10 years of age, when it is already at an advanced stage. Such cases are associated with low EF and high mortality, with death generally occurring within 3 months of diagnosis. These data suggest delayed diagnosis—and more complicated treatment—of DCM in this setting.1
Diagnostic tests that have been proposed for investigating DCM in asymptomatic patients with EB include annual blood tests with complete blood count, albumin levels, total and free carnitine levels, selenium and zinc levels, and cardiac assessment (electrocardiogram and echocardiogram). Patients with symptoms or alterations in any of the above tests should be urgently referred for cardiac assessment. In addition, thyroid hormone measurement, serology, culture of parvovirus and Coxsackie virus, metabolic tests, and myocardial biopsy when appropriate, should be performed.
ConclusionsDCM must be contemplated as a possible complication of EB, especially in patients with recessive dystrophic or junctional EB. Regular testing at a young age is therefore necessary, as is the correction of alterations that have been linked to heart damage in these patients.1 Early detection of anomalies will allow prompt initiation of the necessary treatment, thereby slowing disease progress and prolonging survival.
Ethical DisclosuresProtection of humans and animalsThe authors declare that no tests were carried out in humans or animals for the purpose of this study.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients.
Right to privacy and informed consentThe authors declare that no private patient data appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Batalla A, Vicente A, Bartrons J, Prada F, Fortuny C, González-Enseñat MA. Miocardiopatía en pacientes con epidermólisis ampollosa hereditaria. Actas Dermosifiliogr. 2017;108:544–549.



