








CD30+ primary cutaneous lymphomas comprise a large group of malignant lymphoproliferative disorders that present in the skin without extracutaneous involvement at the time of diagnosis. The annual incidence of these lymphomas is low, at 7–10 cases per 100 million population. Two types, derived from T cells (70%–85%) or B cells (15%–30%), have been identified. Hematologists and oncologists have increasingly recognized the idiosyncrasy of primary cutaneous lymphomas, as reflected in the updated classification of the World Health Organization. However, there remain nuances or small differences to consider when managing these conditions, obliging dermatologists to continue to strive to fully reconcile the various clinical pictures in future reviews of the classification of lymphoid neoplasms. A diagnosis of a primary cutaneous lymphoma is based on clinical, histopathologic, immunophenotypic, and genotypic criteria, particularly evidence of T- or B-cell lymphoid monoclonality in lesions. Also relevant are complementary tests to rule out extracutaneous involvement.
Los linfomas cutáneos primarios son un grupo heterogéneo de procesos linfoproliferativos malignos que se manifiestan inicialmente en la piel sin evidencia de afectación extracutánea en el momento del diagnóstico que presentan una baja incidencia (7–10 casos x 10 6 personas/año). Se dividen en linfomas cutáneos derivados de linfocitos T (70-85%) y de células B (15-30%). El reconocimiento de la idiosinrasia de los linfomas cutáneos primarios por parte de hematólogos y oncólogos es cada vez mayor como queda reflejado en la última actualización de la clasificación Organización Mundial de la Salud, si bien, todavía quedan matices o peculiaridades a considerar en su manejo que obligan a los dermatólogos a seguir trabajando para una plena integración de las diferentes situaciones clínicas que nos plantean en futuras revisiones de la clasificación de las neoplasias linfoides. El diagnóstico de un linfoma cutáneo primario se establece en base a los hallazgos clínicos, histopatológicos, inmunofenotípicos y genotípicos (demostración de monoclonalidad linfoide T o B) de las lesiones cutáneas y en el resultado de las distintas exploraciones complementarias destinadas a descartar una afectación extracutánea.
Primary cutaneous lymphomas (PCLs) are a heterogeneous group of malignant lymphoproliferative processes that initially present on the skin with no evidence of extracutaneous involvement at the time of diagnosis. The annual incidence of this entity is low (7–10 cases per million population),1,2 with T-cell derived PCLs (primary cutaneous T-cell lymphomas PCTCLs) accounting for 70%-85% and B-cell derived ones (primary cutaneous B-cell lymphomas [PCBCLs]) accounting for 15% to 30%.3–5
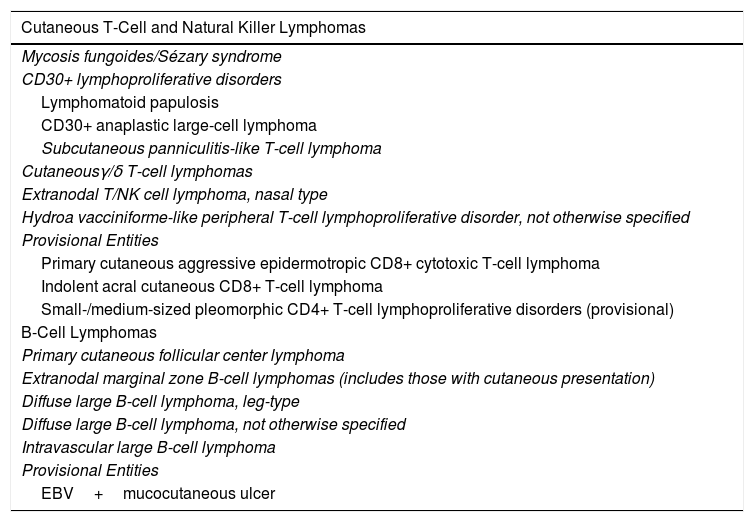
Hematologists and oncologists increasingly recognize the idiosyncrasies of PCLs, as reflected in the most recent update of the World Health Organization (WHO) classification (Table 1).6 However, there are still nuances and special characteristics to take into account in the management of PCLs. Dermatologists should therefore continue in their endeavor to achieve full integration of the different clinical situations in future updates to the WHO classification of lymphoid neoplasms (2018 WHO/EORTC consensus classification for PCLs).1 The most relevant changes from the 2016 WHO classification include: A, Primary cutaneous small-/medium-sized pleomorphic CD4+ T-cell process, the current classification defines as a ‘primary lymphoproliferative process of small/medium sized pleomorphic cells,’ thus losing the condition of true lymphoma; B, Some new provisional entities such as acral CD8+ T-cell lymphoma or mucocutaneous ulcer caused by Epstein-Barr virus (EBV) are defined, whereas other entities such as aggressive epidermotropic CD8+CTCL maintain their provisional character. In the case of PCBCLs, the most recent update of the WHO classification differentiates between follicle center lymphoma and diffuse large BCL, leg type, as separate entities, but it does not consider primary cutaneous marginal zone lymphoma as a separate entity and includes it as part of the extranodal MALT lymphomas (Table 1).
World Health Organization Classification 2016: Known Subtypes of Primary Cutaneous Lymphomas.
| Cutaneous T-Cell and Natural Killer Lymphomas |
|---|
| Mycosis fungoides/Sézary syndrome |
| CD30+ lymphoproliferative disorders |
| Lymphomatoid papulosis |
| CD30+ anaplastic large-cell lymphoma |
| Subcutaneous panniculitis-like T-cell lymphoma |
| Cutaneousγ/δ T-cell lymphomas |
| Extranodal T/NK cell lymphoma, nasal type |
| Hydroa vacciniforme-like peripheral T-cell lymphoproliferative disorder, not otherwise specified |
| Provisional Entities |
| Primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma |
| Indolent acral cutaneous CD8+ T-cell lymphoma |
| Small-/medium-sized pleomorphic CD4+ T-cell lymphoproliferative disorders (provisional) |
| B-Cell Lymphomas |
| Primary cutaneous follicular center lymphoma |
| Extranodal marginal zone B-cell lymphomas (includes those with cutaneous presentation) |
| Diffuse large B-cell lymphoma, leg-type |
| Diffuse large B-cell lymphoma, not otherwise specified |
| Intravascular large B-cell lymphoma |
| Provisional Entities |
| EBV+mucocutaneous ulcer |
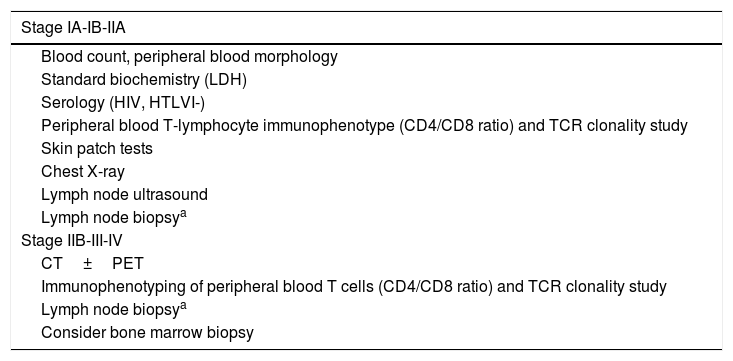
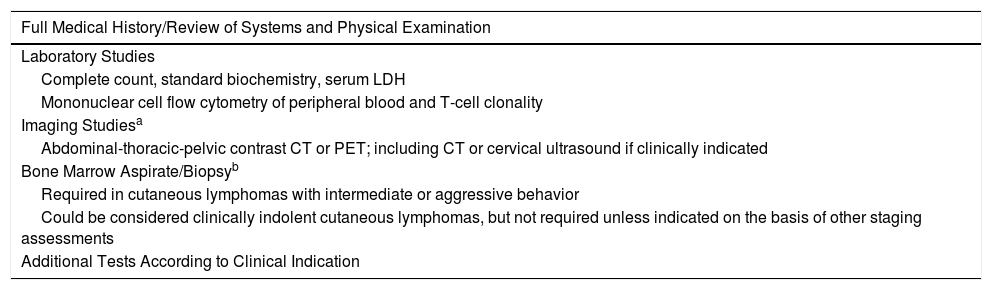
Diagnosis of PCL is usually made on the basis of clinical, pathologic, immunophenotype, and genotype findings (demonstration of T- and B-cell monoclonality) in cutaneous lesions and on the results of different additional tests aimed at ruling out extracutaneous involvement.5,7,8 The additional tests for initial assessment of PCLs are summarized in Tables 2 and 3, while the staging is presented in Tables 4 and 5.
Additional Tests for Diagnosis and Assessment of Mycosis Fungoides/Sézary Syndrome.
| Stage IA-IB-IIA |
|---|
| Blood count, peripheral blood morphology |
| Standard biochemistry (LDH) |
| Serology (HIV, HTLVI-) |
| Peripheral blood T-lymphocyte immunophenotype (CD4/CD8 ratio) and TCR clonality study |
| Skin patch tests |
| Chest X-ray |
| Lymph node ultrasound |
| Lymph node biopsya |
| Stage IIB-III-IV |
| CT±PET |
| Immunophenotyping of peripheral blood T cells (CD4/CD8 ratio) and TCR clonality study |
| Lymph node biopsya |
| Consider bone marrow biopsy |
Additional Tests for Diagnosis and Assessment of Cutaneous T-Cell Lymphomas Other Than Mycosis Fungoides/Sézary Syndrome.
| Full Medical History/Review of Systems and Physical Examination |
|---|
| Laboratory Studies |
| Complete count, standard biochemistry, serum LDH |
| Mononuclear cell flow cytometry of peripheral blood and T-cell clonality |
| Imaging Studiesa |
| Abdominal-thoracic-pelvic contrast CT or PET; including CT or cervical ultrasound if clinically indicated |
| Bone Marrow Aspirate/Biopsyb |
| Required in cutaneous lymphomas with intermediate or aggressive behavior |
| Could be considered clinically indolent cutaneous lymphomas, but not required unless indicated on the basis of other staging assessments |
| Additional Tests According to Clinical Indication |
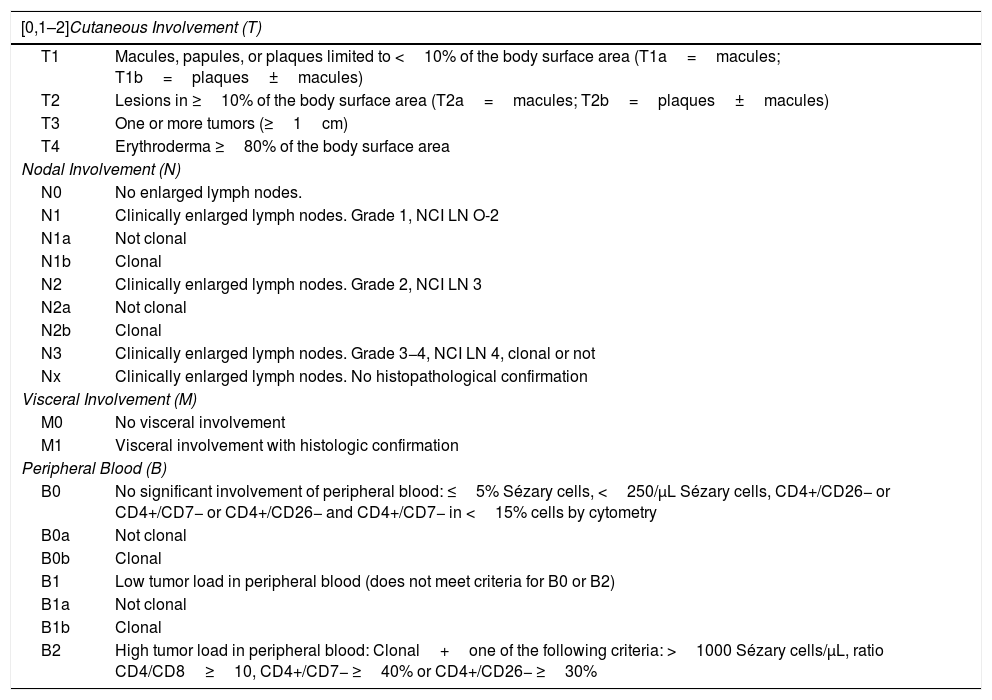
TNM(B) Staging of Mycosis Fungoides/Sézary Syndrome.
| [0,1–2]Cutaneous Involvement (T) | |
|---|---|
| T1 | Macules, papules, or plaques limited to <10% of the body surface area (T1a=macules; T1b=plaques±macules) |
| T2 | Lesions in ≥10% of the body surface area (T2a=macules; T2b=plaques±macules) |
| T3 | One or more tumors (≥1cm) |
| T4 | Erythroderma ≥80% of the body surface area |
| Nodal Involvement (N) | |
| N0 | No enlarged lymph nodes. |
| N1 | Clinically enlarged lymph nodes. Grade 1, NCI LN O-2 |
| N1a | Not clonal |
| N1b | Clonal |
| N2 | Clinically enlarged lymph nodes. Grade 2, NCI LN 3 |
| N2a | Not clonal |
| N2b | Clonal |
| N3 | Clinically enlarged lymph nodes. Grade 3−4, NCI LN 4, clonal or not |
| Nx | Clinically enlarged lymph nodes. No histopathological confirmation |
| Visceral Involvement (M) | |
| M0 | No visceral involvement |
| M1 | Visceral involvement with histologic confirmation |
| Peripheral Blood (B) | |
| B0 | No significant involvement of peripheral blood: ≤5% Sézary cells, <250/μL Sézary cells, CD4+/CD26− or CD4+/CD7− or CD4+/CD26− and CD4+/CD7− in <15% cells by cytometry |
| B0a | Not clonal |
| B0b | Clonal |
| B1 | Low tumor load in peripheral blood (does not meet criteria for B0 or B2) |
| B1a | Not clonal |
| B1b | Clonal |
| B2 | High tumor load in peripheral blood: Clonal+one of the following criteria: >1000 Sézary cells/μL, ratio CD4/CD8≥10, CD4+/CD7− ≥40% or CD4+/CD26− ≥30% |
| T | N | M | B | |
|---|---|---|---|---|
| IA | 1 | 0 | 0 | 0,1 |
| IB | 2 | 0 | 0 | 0,1 |
| II | 1,2 | 1,2 | 0 | 0,1 |
| IIB | 3 | 0−2 | 0 | 0,1 |
| III | 4 | 0−2 | 0 | 0,1 |
| IIIA | 4 | 0−2 | 0 | 0 |
| IIIB | 4 | 0−2 | 0 | 1 |
| IVA1 | 1−4 | 0−2 | 0 | 2 |
| IVA2 | 1−4 | 3 | 0 | 0−2 |
| IVB | 1−4 | 0−3 | 1 | 0−2 |
TNM Staging of Primary Cutaneous T-Cell Lymphomas Other Than Mycosis Fungoides/Sézary Syndrome.
| Cutaneous Involvement (T) |
|---|
| T1 Solitary cutaneous lesion |
| T1a: <5cm |
| T1b: >5cm |
| T2: Multiple lesions grouped in a single anatomical region or 2 contiguous body regions |
| T2a: <15cm total diameter of the affected area |
| T2b: >15 and <30cm total diameter of the affected area |
| T2c: >30cm total diameter of the affected area |
| T3: Multiple generalized lesions |
| T3a: Lesions in >2 nonadjacent anatomical areas |
| T3b: Lesions in ≥3 anatomical regions |
| Nodal Involvement (N) |
| No: No clinical or pathological nodal involvement |
| N1: Peripheral nodal involvement of 1 node in lymphatic drainage territory of the cutaneous lesion |
| N2: Involvement of 2 lymph nodes or 1 peripheral node not in lymphatic drainage territory of the cutaneous lesion |
| N3: Involvement of central lymphatic node(s) |
| Visceral Involvement (M) |
| M0: No visceral involvement |
| M1: Visceral involvement |
PCTCLs include mycosis fungoides (MF), the most frequent subtype, accounting for approximately half these lymphomas, Sézary syndrome (SS), and PCTCLs other than those belonging to MF or SS; this is a heterogeneous group of entities characterized by malignant T-cell proliferation.3,4
Mycosis FungoidesMF is the most frequent subtype of PCL, with an annual incidence of 0.3−0.5 new cases per 100,000 population. It affects adult patients with a 2:1 predilection for men. MF is a monoclonal proliferation of mature memory CD4+/CD45RO+T-helper cells.2
Clinical ManifestationsIn its initial stages, MF remains localized to the skin for years in the form of erythematous-scaly macules or plaques with persistent infiltration, usually at sites corresponding to covered areas of the skin. In the initial phases (stages IA, IB, IIA), the disease has no impact on survival. In a limited percentage of patients (20%), the disease progresses to more advanced stages, developing solitary or multiple nodules or tumors, with a tendency towards ulceration (stage IIB). These lesions usually coexist simultaneously with macules or plaques and/or show specific lymph node and visceral involvement (stage IV) (Fig. 1A-C). Age greater than 60 years, elevated LDH levels, presence of visceral involvement, and histological transformation to large-cell lymphoma are considered factors for poor prognosis in MF.9–11
 lesion in macular phase. B, Plaque-type MF lesion. C, Tumor MF. D, Histology of plaque MF with band-like infiltrate of the superficial and medial reticular dermis with large atypical lymphocytes. Epidermotropic processes may be present. E, Ki67 proliferation marker allows detection of large lymphoid cells. F, Diffuse infiltrate throughout the dermis corresponding to a tumor MF lesion. G, Large cells with pleomorphic nuclei from an MF tumor biopsy. H, Proliferation index >50% in a tumor MF lesion.")
A, Mycosis fungoides (MF) lesion in macular phase. B, Plaque-type MF lesion. C, Tumor MF. D, Histology of plaque MF with band-like infiltrate of the superficial and medial reticular dermis with large atypical lymphocytes. Epidermotropic processes may be present. E, Ki67 proliferation marker allows detection of large lymphoid cells. F, Diffuse infiltrate throughout the dermis corresponding to a tumor MF lesion. G, Large cells with pleomorphic nuclei from an MF tumor biopsy. H, Proliferation index >50% in a tumor MF lesion.
Histopathological study of initial macules shows an infiltrate in the superficial dermis, formed of small cerebriform lymphocytes that are often distributed along the basal layer of the epidermis (epidermotropism) with no spongiotic changes. Progression of the lesions to plaques gives rise to a denser band-like infiltrate in the papillary dermis, and stronger epidermotropism (intraepidermal Pautrier microabscesses). The tumors are characterized by dense monomorphic infiltrates of atypical nodular or diffuse lymphocytes, involving the full thickness of the dermis. The presence of large cells (usually CD30+) is frequent and when these account for more than 25% of the infiltrate, large-cell transformation is considered to have occurred (Fig. 1D-H). The usual phenotype is CD2+, CD3+, βF1+, CD4+, CD8–, CD45RO with a loss of mature T-lymphocyte markers (CD7, CD, or CD5). A cytotoxic phenotype (CD8+, TIA1+ or γδ+) in MF is not usually associated with differences in clinical presentation, prognosis, or progression. Clonality study by polymerase chain reaction detects monoclonal T-cell receptor (TCR) reordering in most cases.3
Pathogenic MechanismsPatients with MF/SS have complex, heterogeneous, nonspecific genomic abnormalities in the genes implicated in T-cell activation, apoptosis, chromatin remodeling, or response to DNA damage, as well as cell cycle regulating genes (TP53, PLCG1, CARD11, STAT5B, NFKB2, IL6, ITPR1, RASA2, TNFRSF10A, FASN, ZEB1, DNMT3A, or KMT2C). Specifically, activation of signaling pathways such as STAT, NOTCH1, or β-catenin (by NF-kB) appear to be key in this process.12–20
Variants of Mycosis FungoidesFolliculotropic Mycosis FungoidesFolliculotropic MF (FMF) is a particular clinicopathologic variant characterized by preferential tropism for the hair follicle epithelium. It can be associated with follicular mucinosis. The lesions are usually located on the face, neck, and trunk, and are manifest as grouped follicular papules, cysts, comedones, areas of alopecia, and/or pseudotumoral lesions (Fig. 2A and B). Overall survival at 10 years is approximately 82% compared with 91% for the classic form, and 41% and 90%, respectively, at 15 years. However, recently, the presence of different subgroups of FMF has been proposed: an early or ‘superficial’ form with a small prominent neoplastic component with a prognosis similar to classic MF in its initial stages, and a ‘deep’ variant with a more aggressive course.21,22 Syringotropic MF is the term used for those forms of MF with exclusive or predominant involvement of the eccrine glands. The lesions usually comprise infiltrated, hyperpigmented and anhidrotic plaques and, often, alopecia. Syringotropic MF can develop either in isolation or associated with FMF (adnexotropic MF).
Pagetoid reticulosis. B, Histology of an incipient or superficial papular FMF lesion that exhibits lymphoid folliculotropism and follicular mucinosis processes.")
Pagetoid reticulosis (PR) is a localized variant of MF, characterized clinically by one or several hyperkeratotic plaques, generally in distal areas of the limbs, and histopathologically by strong epidermotropism. Neoplastic cells present a CD8+ or CD4+ phenotype and often, they are CD30+. They should be differentiated from other subtypes of aggressive PCTCLs with prominent epidermotropism, as PR follows an indolent clinical course, with localized radiotherapy as the treatment of choice.1
Granulomatous slack skinGranulomatous slack skin is a particular and uncommon variant of MF characterized clinically by slow and progressive development of redundant skin in the large folds (axillae, groin). Histologically, a dense infiltrate is observed with granulomatous features, with dense dermal lymphoid infiltrates and abundant multinucleated giant cells with elastophagocytosis and elastolysis. In addition, infiltration of superficial layers is usually observed with atypical lymphoid cells and a variable degree of epidermotropism.1
Sézary syndromeTraditionally, SS is characterized by erythroderma, generalized swollen lymph nodes, and more than 1000/mm3 (or >10%) atypical circulating mononuclear cells with cerebriform nucleus (Sézary cells). It is observed almost exclusively in elderly adults and has a certain predilection for men. Along with diffuse erythroderma (erythema that affects >80% of body surface) associated with intense pruritus, it is usually accompanied by ectropion, palmoplantar keratoderma, nail dystrophy, and alopecia (Fig. 3). The circulating Sézary cells are CD4+, CD7-, CD26- with a CD4+/CD8+ T-cell ratio >10. Evidence of clonal expansion of CD4+/CD7− ≥40% or CD4+/CD26− ≥30% would also be considered sufficiently strong for diagnosis. Cutaneous histopathological findings in SS are similar to those observed in MF, although epidermotropism is usually weaker. The disease usually follows an aggressive course, and 5-year survival does not exceed 30%–40%.7,10,23
Treatment of mycosis fungoides and sézary syndrome
Patients with stage IA-IIA disease should be initially treated with therapies targeting the skin, such as topical corticosteroids or phototherapy (psoralen-UV-A [PUVA] therapy or narrow-band UVB therapy).24,25 Topical cytostatic agents (mechlorehamine or carmustine) can achieve therapeutic responses in more than 50% of patients, although their use is limited by poor tolerance (irrative dermatitis).26 Other topical treatments include gel bexarothen (not reimbursed), resiquimod (unapproved toll-like receptor agonist), and occasionally calcineurin inhibitors (also unapproved) or photodynamic therapy.27,28 In those patients with disseminated infiltrated plaques, or in patients who are refractory to the aforementioned treatments, oral bexarotene29 or interferon-α30 are usually prescribed as monotherapy, or, alternatively, combinations of bexarotene, inferferon-α, or PUVA therapy may be used.24 Total skin electron radiation is an effective therapeutic alternative in some patients with MF in stages IB or IIA (Fig. 4).31–33 In FMF, the best results appear to be achieved with the use of PUVA associated with bexaroten or interferon-α or with total skin electron radiation.34

In patients with single or localized tumors (stage IIB), localized radiotherapy is used, while in the case of multiple lesions, gemcitabine or doxorubicin are used in monotherapy.3,35,36 Other treatments that modify biological response, such as histone deacetylase inhibitors (vorinostat, romidepsin), used both for advanced and early disease, in monotherapy or in combination with other treatments, could be of use, although they are still not approved in the European Union.37 Polychemotherapy regimens (CHOP) are only indicated in patients with lymph node and/or visceral involvement (stage IV) who are refractory to the aforementioned therapies or in the context of treatment prior to hematopoietic stem cell transplantation. Recently, brentuximab vedotin (monoclonal antibody to the CD30 antigen) and mogamulizumab (monoclonal antibody to the CCR4 receptor) have been approved in patients with advanced MF and SS (Fig. 4).38,39
Extracorporeal photopheresis is considered the initial treatment of choice for SS after diagnosis.40 It can be associated with interferon-α, bexarotene, full body radiation, or PUVA therapy. Methotrexate, prednisone, and chlorambucil are classic therapies in SS, although their efficacy is relatively low. In refractory patients, management of advanced SS is not notably different to that of advanced MF, although some drugs such as alemtuzumab (monoclonal antibody to CD52 antigen) and mogamulizumab could have a more favorable efficacy/toxicity profile in patients refractory to SS.41 Mogamulizumab has demonstrated better therapeutic response in the blood compartment (unlike brentuximab, which appears to have better efficacy in the cutaneous compartment)(Fig. 4).23,24,38,39
Finally, in patients with advanced MF/SS, the possibility of hematopoietic stem cell transplantation should be considered.42 Alternatively, such patients could be candidates for clinical trials, given that several molecules are in development aimed at both controlling immune response (anti-PD1 or anti-PDL1 monoclonal antibodies) and enhancing monoclonal antibody-mediated cytotoxicity (KIRDL2).43
Primary Cutaneous CD30+ Lymphoproliferative DisordersPrimary cutaneous CD30+ lymphoproliferative disorders are a group of cutaneous lymphoproliferative disorders that have in common expression of the lymphoid CD30 activation antigen by neoplastic cells. The range of entities includes lymphomatoid papulosis (LP) and CD30+ anaplastic large-cell lymphoma (CD30+ALCL). Prognosis is usually good, with mean survival in excess of 90% at 5 years.1
Lymphomatoid papulosisClinical manifestationsLP is characterized by the recurrent and chronic development of papules or nodules that spontaneously involute, often leaving residual scars. The lesions are usually erythematous papules, with a tendency to ulceration and central necrosis, and of variable size, number and distribution (isolated, grouped, or disseminated lesions) usually located on the trunk and extremities (Fig. 5A). Between 5% and 20% of patients may show an association with another neoplastic process, usually hematologic (Hodgkin lymphoma) or MF.44
. B, Dermal infiltrate formed of atypical lymphocytes and rich in eosinophils from a papular LP lesion. C, Large-cell lymphocytes with nuclear pleomorphism in an LP lesion. D, Expression of CD30 antigen in large lymphocytes with an activated appearance in LP.")
A, Papules at different stages of development corresponding to lymphomatoid papulosis (LP). B, Dermal infiltrate formed of atypical lymphocytes and rich in eosinophils from a papular LP lesion. C, Large-cell lymphocytes with nuclear pleomorphism in an LP lesion. D, Expression of CD30 antigen in large lymphocytes with an activated appearance in LP.
LP shows a lymphoid infiltrate of perivascular and interfacial wedge-shaped arrangement in the dermis, occasionally spreading to subcutaneous tissue. Atypical large lymphoid cells, which express CD30 antigen, similar to Reed-Sternberg cells in Hodgkin lymphoma, can be observed in this infiltrate. Likewise, a dense accompanying inflammatory infiltrate can be observed with abundant polymorphonuclear neutrophils and eosinophils. Histopathological findings observed may require differential diagnosis with lichenoid pityriasis or even with arthropod bites. The neoplastic cells express the T-helper phenotype (CD3+, CD4+, CD8−) with the presence of groups of atypical CD30+ large cells (Fig. 5B-D).44
In recent years, different histopathological variants have been reported. Along with the classic type A (histiocytic), type B (similar to MF), or type C (similar to PCTCL-CD30+) forms,44 variants with cytotoxic phenotype (type D) have been described.45 These processes may resemble a primary cutaneous aggressive epidermotropic cytotoxic T-cell lymphoma. Variants with angioinvasive histology (type E),46 which are usually manifest clinically with ulcerous-necrotic lesions, predominantly folliculotropic, granulomatous, forms, etc, have also been described. These variants do not follow a distinct clinical course, although they may present diagnostic difficulties to differentiate from other lymphoproliferative disorders. Genetic mutations specific to LP have not been described, although recently reordering of the DUSP22 gene with IRF4 at the 6p25.3 locus has been reported in a limited number of cases.47
Assessment and treatmentDiagnosis of PL is established according to clinical-pathologic features of the cutaneous lesions and its self-resolving character. Treatment of LP is determined by the extent and characteristics of the lesions. Often, no treatment is required. In cases of extensive, necrotic lesions, or lesions with a tendency to cause scarring, treatment with low-dose oral methotrexate (7.5 to 15 mg/week or even lower doses), phototherapy (UVB or PUVA therapy), or interferon-α can be prescribed. Exceptionally, treatment with brentuximab could be considered.38,44
Primary cutaneous CD30+ anaplastic large cell lymphomaClinical manifestationsPrimary cutaneous CD30+ anaplastic large-cell lymphoma presents characteristically as a solitary nodule or multiple nodules of variable size in groups or at different anatomical sites; these lesions are fast-growing and with a tendency to ulcerate (Fig. 6A). For staging, the TNM classification for different PCL of the MF/SS group is applied (Table 5). Approximately 10% of patients can develop extracutaneous involvement, generally of locoregional lymph nodes.44
Histopathology
Histologically, this process is characterized by diffuse dermal infiltration formed of large atypical lymphoid cells with abundant cytoplasm, rounded-oval nuclei, and prominent nucleoli. These cells have a CD4+/CD45RO+T-cell phenotype with intense expression of CD30 antigen (>75% of neoplastic cells) (Fig. 6B-D). Expression of CD56 or cytotoxic markers is not exceptional (TIA-1, granzyme B) but without any prognostic implications.44
In CD30+PCTCL, t(2;5)(p23;q35) chromosomal translocation is not detected; this finding characterizes nodal origin anaplastic lymphomas. Likewise, expression of its corresponding fusion protein NPM/ALK (anaplastic lymphoma kinase) associated with this translocation is not detected. The EMA antigen is also not expressed and these cells are CD15- unlike those of nodal anaplastic lymphomas and Hodgkin lymphoma. In approximately 25% of cases, DUSP22/IRF4 translocation can be detected.44
Prognosis and treatmentThis entity usually has a good prognosis, even in those cases with secondary locoregional lymph node involvement, with 5-year survival rates greater than 75% to 90%. Conservative treatment with surgery or radiotherapy is often recommended in solitary lesions, or else methotrexate, interferon, or monochemotherapy in those cases with localized lesions. In the event of extensive multicentric cutaneous involvement, treatment with brentuximab should be considered, and only exceptionally with other more aggressive alternatives (polychemotherapy).44
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Pujol RM and Gallardo F. Linfomas cutáneos. Parte I: micosis fungoide, síndrome de Sézary y proliferaciones linfoides cutáneas CD30 positivas. Actas Dermosifiliogr. 2021;112:14–23.



